一类新型萘酚类化合物的制备及其在癌症治疗方面的应用的制作方法
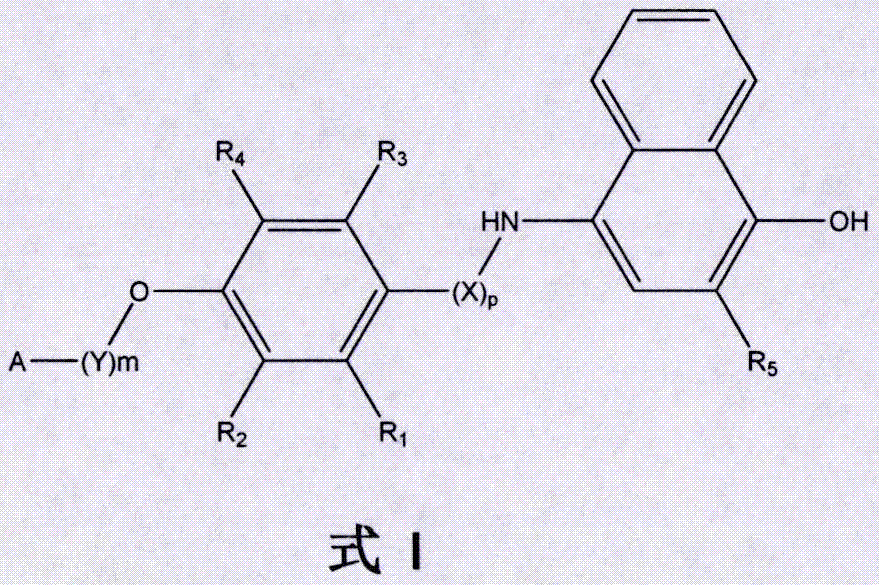
本发明涉及医药生物领域,更具体地,涉及一类新型萘酚类化合物的制备及其在癌症治疗方面的应用。
背景技术:
2017年国家癌症中心发布的中国最新癌症数据显示,在中国,每年新发癌症病例达429万,占全球新发病例的20%,死亡281万例(中国肿瘤临床与康复,2017(5):574-574)。2017年中国cfda批复上市的5个抗癌新药没有一个创新靶点,中国原创的靶向新药寥寥无几,总体反映了国内靶向原创药物的研发水平落后于欧美。如何在药物研发方面做到“双创”(生物靶点是与疾病相关的首次用于临床治疗的新靶点和全新化合物)是考验我们中国医药研发实力的试金石。寻求新的靶点和有潜力的药物先导化合物,在特定的肿瘤治疗领域有所突破,是医药研发科研人员的当务之急。stat3-jak信号传导通路对肿瘤细胞的生长有正向调控的作用,stat3蛋白作为治疗癌症的生物学靶点近十年备受青睐,截止2017年,美国fda批准在临床测试的stat3信号传导通路抑制类抗癌药物有30余种(johnsonde,etal.,naturereviewsclinicaloncology,2018,15(4):234)。stat3抑制剂类抗癌靶向药物有靶点新和抗癌谱宽等特点,近期的临床测试结果显示了此类药物在未来肿瘤临床治疗方面具有巨大的开发潜力和广阔的市场空间。申请人发现的smbaa002和smbaa007等此类萘酚化合物属于stat3抑制剂,该类化合物抑制stat3活化的机制清晰、抑制肿瘤细胞生长的效果显著。本化合物由河南省锐达医药科技有限公司研发。
此萘酚类化合物因其独特的结构特征,它可以通过结构中的极性键和原子与生物体内与肿瘤疾病相关的蛋白位点相结合,并与受体发生氢键作用。另一方面,结构中的芳香环与肿瘤疾病相关的酶和受体形成芳环堆叠作用,从而达到抑制肿瘤细胞增殖的目的。
技术实现要素:
本发明主要解决的技术问题是提供了一类具有式i结构的新型萘酚类化合物的制备及对肿瘤细胞抑制作用的应用。
为实现上述发明目的,本发明提供了以下技术方案:
一种具有式i结构的萘酚类化合物,
其中,
r1、r2、r3、r4各自独立的选自:氢、卤素、硝基、烷基、氰基、芳基;
r5选自:-h、-oh;
p=0或1,
x为-ch2-、-(ch2)2-、-co-、-ch2-co-,-(ch2)2-co-等;
m=0或1,
y为-ch2-、-(ch2)2-、-co-、-ch2-co-、-(ch2)2-co-等;
a选自:
本发明所用的术语“卤素”是指氟、氯、溴或碘,优选的卤素基团为氟、氯或溴。进一步,更具体的来说,本发明所述的萘酚类化合物是以下化合物之一:
本发明同时还提供了上述式i萘酚类化合物的制备方法,包括如下步骤:
化合物通式i(其中x=-co-、-ch2-co-、-(ch2)2-co-、y=-ch2-、-(ch2)2-、r5=-h)可以由流程1所示路线合成,原料经过mitsunobu反应形成醚化中间体,再经强碱溶液水解生成对应羧酸,羧酸纯化后进行酰化反应生成对应酰氯,最后在碱的环境中合成目标化合物。
流程1
1a:x=-co-,y=-ch2-,e=-ch2-,r1=h,r2=h,r3=h,r4=h
1b:x=-co-,y=-ch2-,e=-o-,r1=h,r2=h,r3=h,r4=h
1c:x=-co-,y=-ch2-,e=-(ch2)2-,r1=h,r2=h,r3=h,r4=h
1d:x=-co-,y=-(ch2)2-,e=-(ch2)2-,r1=h,r2=h,r3=h,r4=h
1e:x=-co-,y=-(ch2)2-,e=-ch2-,r1=h,r2=h,r3=h,r4=h
1f:x=-co-,y=-(ch2)2-,e=-o-,r1=h,r2=h,r3=h,r4=h
对于x、y和e的具体基团包括上述1a,1b,1c,1d,1e,1f但不限于这些基团/化合物,还可以是其他的本领域技术人员容易理解想到采用该流程1进行合成的化合物。以下流程2-3中的特别化合物及x、y、e的限定与上述情况相同,包括但不限于这些具体的化合物成分。对于权利要求书中的限定的合成工艺/流程的情况应作同样的理解,不应被视为限定,更不能限定为流程中具体的化合物。
化合物通式i(其中x=-ch2-、-(ch2)2-、y=-ch2-、-(ch2)2-、r5=-h)可以由流程2所示路线合成,原料经过mitsunobu反应形成醚化中间体,最后在强碱的环境中合成目标化合物。
流程2
3a:x=-(ch2)2-,y=-ch2-,e=-ch2-,r1=h,r2=h,r3=h,r4=h
3b:x=-(ch2)2-,y=-ch2-,e=-o-,r1=h,r2=h,r3=h,r4=h
3c:x=-(ch2)2-,y=-ch2-,e=-(ch2)2-,r1=h,r2=h,r3=h,r4=h
3d:x=-(ch2)2-,y=-(ch2)2-,e=-ch2-,r1=h,r2=h,r3=h,r4=h
3e:x=-(ch2)2-,y=-(ch2)2-,e=-o-,r1=h,r2=h,r3=h,r4=h
3f:x=-(ch2)2-,y=-(ch2)2-,e=-(ch2)2-,r1=h,r2=h,r3=h,r4=h
化合物通式i(其中x=-co-、-ch2-co-、-(ch2)2-co-、y=-ch2-、-(ch2)2-,r5=-oh)可以由流程3所示路线合成,原料为经过流程1合成的酰胺中间体,再经双氧水氧化生成醌式中间体,最后在稀碱中经迈克尔加成合成目标化合物。
流程3
2a:x=-co-,y=-ch2-,e=-ch2-,r1=h,r2=h,r3=h,r4=h
2b:x=-co-,y=-ch2-,e=-o-,r1=h,r2=h,r3=h,r4=h
2c:x=-co-,y=-ch2-,e=-(ch2)2-,r1=h,r2=h,r3=h,r4=h
2d:x=-co-,y=-(ch2)2-,e=-ch2-,r1=h,r2=h,r3=h,r4=h
2e:x=-co-,y=-(ch2)2-,e=-o-,r1=h,r2=h,r3=h,r4=h
2f:x=-co-,y=-(ch2)2-,e=-(ch2)2-,r1=h,r2=h,r3=h,r4=h
本发明的另一目的是提供一种用于抑制肿瘤细胞增殖的化合物,该化合物含有式i萘酚类化合物或其生物学可接受的盐,作为活性成分。可以是上述方法合成的萘酚类化合物的情况中的任意一种,可以仅含有其中一种,也可以是含有其中多种复合应用的情况。
进一步,生物学可接受的盐是指式i化合物与乙酸、二氢乙酸、苯甲酸、柠檬酸、山梨酸、丙酸、草酸、富马酸、马来酸、盐酸、苹果酸、磷酸、亚硫酸、硫酸、香草酸、酒石酸、抗坏血酸、硼酸、乳酸和乙二胺四乙酸中的至少一种形成的盐。
本发明的目的是寻找具有高stat3抑制作用且具有较低毒性的新化合物。
本发明还涉及上述的具有式i结构的新型萘酚类化合物、其药学上可接受的盐、所述衍生物的溶剂合物、或者所述盐的溶剂合物在制备用于治疗或辅助治疗和/或预防哺乳动物的肺癌、结肠癌、乳腺癌以及白血病的药物中的用途。具体地,所述哺乳动物为人类。
本发明还涉及上述的具有式i结构的新型萘酚类化合物、其药学上可接受的盐、所述衍生物的溶剂合物、或者所述盐的溶剂合物在制备用于治疗或辅助治疗和/或预防哺乳动物由stat3介导的肿瘤或由stat3驱动的肿瘤细胞增殖和迁移药物中的用途。具体地,所述哺乳动物为人类。
本发明一个方面涉及上述的具有式i结构的新型萘酚类化合物、其药学上可接受的盐、所述衍生物的溶剂合物、或者所述盐的溶剂合物制备用于治疗和/或预防哺乳动物中与stat3细胞信号传导相关疾病的药物中的用途。具体地,所述哺乳动物为人类。
根据本发明,完全可以预期本发明化合物可用于治疗stat3异常高表达而引起的肿瘤。stat3相关的肿瘤包括肺癌、乳腺癌、结直肠癌、白血病等其他所有癌症。
本发明使用的各种术语和短语具有本领域技术人员公知的一般含义,即便如此,本发明仍然希望在此对这些术语和短语作更详尽的说明和解释,提及的术语和短语如有与公知含义不一致的,以本发明所表述的含义为准。
在本发明合成式i化合物的方法中,反应所用的各种原材料是本领域技术人员根据已有知识可以制备得到的,或者是可以通过文献公知的方法制得的,或者是可以通过商业购得的。以上反应方案中所用的中间体、原材料、试剂、反应条件等均可以根据本领域技术人员已有知识做适当改变的。
附图说明
图1是通式i附图。
图2是化合物1a的盐酸盐核磁图谱。
图3是化合物2a核磁图谱。
图4是实施例3中化合物smbaa002与stat3蛋白sh2功能域虚拟分子对接的实验结果,化合物smbaa002呈现在stat3蛋白sh2功能域作用界面上,非极性氢原子(h)均无标记,在stat3蛋白sh2区间上面,与smbaa002分子相互作用的关键氨基酸有:赖氨酸591和精氨酸609和丝氨酸611、613、636和谷氨酸612、638,分别用lys591、arg609、ser611、glu612、ser613、ser636和glu638标记;stat3蛋白sh2区间的β折叠,α螺旋和无规则卷曲分别用缓直带,螺旋带和细管表示。虚拟对接的结果显示,化合物smbaa002与stat3蛋白sh2功能域的磷酸化作用位点arg609、特异性氨基酸glu638和药物疏水选择空腔均有强相互作用,推断该化合物为作用在stat3磷酸化作用位点的stat3信号传导抑制剂。
图5是化合物smbaa002(2a)诱导乳腺癌、肺癌癌细胞凋亡mtt实验,图中mtt细胞实验的结果以ic50(μmol/l)值进行表征。
图6是化合物smbaa007(1a)乳腺癌、白血病、结肠癌和肺癌癌细胞凋亡mtt实验,图中mtt细胞实验的结果以ic50(μmol/l)值进行表征。
图7是化合物smbaa002(2a)蛋白免疫印迹的结果。蛋白免疫印迹实验的结果以将电泳分离后的细胞总蛋白质从凝胶转移到固相支持物膜上,根据抗原抗体特异原理,分别通过stat3、p-stat3、β-actin抗体检测相应蛋白表达量。结果如图,可以看到在药物作用下,随药物浓度增加,hcc827表达stat3、β-actin蛋白量不变,而p-stat3表达量呈下降趋势,化合物smbaa002(2a)明显抑制p-stat3的表达。
图8是化合物smbaa007(1a)蛋白免疫印迹的结果。蛋白免疫印迹实验的结果以将电泳分离后的细胞总蛋白质从凝胶转移到固相支持物膜上,根据抗原抗体特异原理,分别通过stat3、p-stat3、β-actin抗体检测相应蛋白表达量。结果如图,可以看到在药物作用下,随药物浓度增加,hcc827表达stat3、β-actin蛋白量不变,而p-stat3表达量呈下降趋势,化合物smbaa007(1a)明显抑制p-stat3的表达。
图9是化合物smbaa007(1a)反转录聚合酶链式反应试验(pcr)结果。利用半定量rt-pcr测定相关抗调亡基因表达情况,验证不同药物浓度作用下hcc827的生理情况;分别将不同浓度作用下的hcc827,通过rna样品抽提、测定rna含量、样品反转录cdna、pcr扩增β-actin、survivin表达;分别对比抗调亡基因在不同药物作下表达情况,可以看出化合物smbaa007(1a)对survivin表达有明显抑制作用。
以上所述化合物部分信息见下表:
具体实施方式
下面通过具体的制备实施例和模拟以及生物学试验例进一步描述本发明,但是,应当理解为,这些实施例和试验例仅仅是用于更详细具体地说明之用,而不应理解为用于以任何形式限制本发明。
本发明对试验中所使用到的材料以及试验方法进行一般性和/或具体的描述。虽然为实现本发明的目的所使用的许多材料和操作方法是本领域公知的,但是本发明仍然在此作尽可能详细描述。本领域技术人员清楚,在下文中,如果未特别说明,本发明所用的材料和操作方法是本领域公知的。
在本发明中,除非另外说明,其中:(i)温度以摄氏度(℃)表示,操作在室温环境下进行;更具体地,所述室温是指20-30℃;(ii)有机溶剂用常用干燥方法干燥,溶剂的蒸发使用旋转蒸发仪减压蒸发,浴温不高于50℃;(iii)反应过程用薄层色谱(tlc)跟踪;(iv)终产物具有满意的质子核磁共振(1h-nmr)和质谱(ms)数据。
实施例1:化合物1a-1f的合成
1a:x=-co-,y=-ch2-,e=-ch2-,r1=h,r2=h,r3=h,r4=h
1b:x=-co-,y=-ch2-,e=-o-,r1=h,r2=h,r3=h,r4=h
1c:x=-co-,y=-ch2-,e=-(ch2)2-,r1=h,r2=h,r3=h,r4=h
1d:x=-co-,y=-(ch2)2-,e=-(ch2)2-,r1=h,r2=h,r3=h,r4=h
1e:x=-co-,y=-(ch2)2-,e=-ch2-,r1=h,r2=h,r3=h,r4=h
1f:x=-co-,y=-(ch2)2-,e=-o-,r1=h,r2=h,r3=h,r4=h
具体合成方法,以化合物1a为例:
n-(4-羟基萘基)-4-[2-(n-哌啶)-乙氧基]苯酰胺
步骤1.4-[2-(n-哌啶)-乙氧基]-苯甲酸甲酯(3)
将n-羟乙基哌啶(6.45g,50mmol)液体,加入无水四氢呋哺80ml,顺序加入尼泊金甲酯(7.60g,50mmol)、三苯基膦(14.41g,55mmol)后氮气保护下冰盐浴降温至0℃开始滴加偶氮二甲酸二异丙酯(11.12g,55mmol),温度会明显升高,体系颜色渐渐变浅黄色透明,控制温度不高于30℃,搅拌反应4h,tlc检测反应完全后,加入2n盐酸调至ph=4,用50ml乙酸乙酸萃取,分液后取水层,水层用碳酸氢钠水溶液调至ph=10后,加入50ml乙酸乙酯萃取,有机层干燥浓缩后得浅黄色油状化合物10.1g(收率76%)。
1hnmr:(300mhz,cdcl3)δ7.90(d,j=8.8hz,2h),6.84(d,j=8.8hz,2h),4.08(t,j=6.0hz,2h),3.81(s,3h),2.71(t,j=6.0hz,2h),2.53-2.36(m,4h),1.61-1.46(m,4h),1.38(dd,j=10.7,5.5hz,2h)。
步骤2.4-[2-(n-哌啶)-乙氧基]-苯甲酸(4)
将4-[2-(n-哌啶)-乙氧基]-苯甲酸甲酯(3)(10g,38mmol),加入50%氢氧化钠水溶液12ml,加入四氢呋喃50ml,升温到50℃反应10h,tlc检测反应完全后,50℃旋蒸浓缩掉四氢呋喃,冷却至室温后,用1n盐酸调ph,直至大量白色固体出现,监测ph约为4~5,过滤得到白色固件粉末,真空干燥箱干燥得白色固体化合物8.1g(收率85.6%)。
步骤3.4-[2-(n-哌啶)-乙氧基]-苯甲酰氯(5)
将4-[2-(n-哌啶)-乙氧基]-苯甲酸(4)(8g,32mmol),加入二氯甲烷50ml,向此悬浊液中滴加氯化亚砜20ml,控制温度不高于40℃,滴加完毕升温至40℃回流反应12h,体系逐渐变为浅黄色透明溶液。体系在50℃用真空水泵浓缩至无液体滴出,得到白色固件粉末9g(盐酸盐形式,收率按100%计算)。
步骤4.n-(4-羟基萘基)-4-[2-(n-哌啶)-乙氧基]苯酰胺(1a)
将4-氨基-1-萘酚盐酸盐(6g,30mmol),加入吡啶60ml,室温(25℃)搅拌30min,体系变为紫黑色溶液,氮气保护下降温至0℃向体系中滴加4-[2-(n-哌啶)-乙氧基]-苯甲酰氯(5)(步骤3所得,32mmol),滴加完毕升至室温反应过夜,tlc检测反应完全,浓缩掉吡啶,加入80ml水搅拌直至体系固件分散,过滤得到棕色固件粉末,用30ml乙酸乙酯打浆30min后过滤后干燥得棕色固件粉末8.9g(1a的盐酸盐,收率69.5%)。
1hnmr:(300mhz,dmso-d6)δ10.71(s,1h),10.27(s,1h),10.09(s,1h),8.17(dd,j=6.7,2.6hz,1h),8.09(d,j=8.5hz,2h),7.84-7.75(m,1h),7.53-7.43(m,2h),7.29(d,j=8.0hz,1h),7.14(d,j=8.7hz,2h),6.92(d,j=8.0hz,1h),4.53(t,j=4.8hz,2h),3.61-3.43(m,4h),3.01(s,2h),1.81(d,j=4.9hz,4h),1.70(d,j=8.8hz,1h),1.40(t,j=18.4hz,1h)。
实施例2:化合物2a-2f的合成
2a:x=-co-,y=-ch2-,e=-ch2-,r1=h,r2=h,r3=h,r4=h
2b:x=-co-,y=-ch2-,e=-o-,r1=h,r2=h,r3=h,r4=h
2c:x=-co-,y=-ch2-,e=-(ch2)2-,r1=h,r2=h,r3=h,r4=h
2d:x=-co-,y=-(ch2)2-,e=-ch2-,r1=h,r2=h,r3=h,r4=h
2e:x=-co-,y=-(ch2)2-,e=-o-,r1=h,r2=h,r3=h,r4=h
2f:x=-co-,y=-(ch2)2-,e=-(ch2)2-,r1=h,r2=h,r3=h,r4=h
具体合成方法,以化合物2a为例:
n-(3,4-二羟基萘基)-4-[2-(n-哌啶)-乙氧基]苯酰胺
步骤1.
(e)-n-(4-oxonaphthalen-1(4h)-ylidene)-4-(2-(piperidin-1-yl)ethoxy)benzamide(3)
将实施例1方法合成的化合物n-(4-羟基萘基)-4-[2-(n-哌啶)-乙氧基]苯酰胺(1a)(1g,2.6mmol)粉末,加入30%双氧水中,室温(25℃)搅拌反应48h,体系由棕色悬浊液慢慢变为红棕色溶液,后颜色慢慢变浅为红色溶液,加入二氯甲烷10mlx2萃取两遍,有机层浓缩得黄色固体430mg(收率43%)。
1hnmr:(300mhz,dmso-d6)δ8.17(dd,j=6.7,2.6hz,1h),8.09(d,j=8.5hz,2h),7.84-7.75(m,1h),7.53-7.43(m,2h),7.29(d,j=8.0hz,1h),7.14(d,j=8.7hz,2h),6.92(d,j=8.0hz,1h),4.08(t,j=6.0hz,2h),2.71(t,j=6.0hz,2h),2.53-2.36(m,4h),1.61-1.46(m,4h),1.38(dd,j=10.7,5.5hz,2h)。
步骤2.n-(3,4-二羟基萘基)-4-[2-(n-哌啶)-乙氧基]苯酰胺(2a)
将(e)-n-(4-oxonaphthalen-1(4h)-ylidene)-4-(2-(piperidin-1-yl)ethoxy)benzamide(3)(400mg,1mmol),加入10%碳酸氢钠钠水溶液5ml,室温搅拌反应72h,tlc检测生成大极性化合物,停止搅拌,静置过夜,有土黄色固体沉淀,过滤后用少量二氯甲烷冲洗,干燥得土黄色固体137mg(收率33%)。
1hnmr:(300mhz,dmso-d6)δ10.11(s,1h),9.79(s,1h),9.51(s,1h),8.17(dd,j=6.7,2.6hz,1h),8.09(d,j=8.5hz,2h),7.89-7.82(m,1h),7.47(d,j=8.0hz,1h),7.31(d,j=8.0hz,1h),7.13(d,j=8.5hz,2h),6.71(s,1h),4.10(t,j=6.0hz,2h),2.91(t,j=6.0hz,2h),2.55-2.38(m,4h),1.61-1.46(m,4h),1.38(dd,j=10.7,5.5hz,2h)。
实施例3:分子对接(docking)实验
方法:为了验证化合物smbaa002与stat3蛋白的相互作用机制,发明人用stat3sh2区的磷酸化络氨酸(py-705)结合区域作为计算机虚拟模拟(docking)的蛋白质模板,虚拟对接区域主要集中在磷酸化酪氨酸作用位点arg609和lys591附近区域。stat3sh2的结构坐标取于蛋白质结构数据库(pdbdatabank,id:1bg1)。分子对接(docking)的方法:所有的计算机配位模拟(docking)的实验均在sybylx2.1.1的操作平台上完成,所用的计算机配位模拟(docking)的工具为sueflexdock。依据所选的位点(主要包括磷酸化酪氨酸作用位点arg609和lys591)进行计算,确定势能面(potentialgradient)并作计算机配位模拟(docking)的实验。依据模拟(docking)的分数(score)和构象及相互作用进行分析。
实施例4:诱导肺癌癌细胞凋亡mtt的实验
方法:收集对数生长期hcc827细胞,计数,调整细胞悬液浓度为50000个/ml,每孔加入100ul细胞悬液即每孔5000个细胞;hcc827细胞加化合物处理,终浓度分别为0.1、0.3、1、3、10、30、100、300(μmol/l)几个梯度,并继续培养48h;药物处理后,每孔加入噻唑蓝试剂50μl(1mg/ml),37℃孵育4h,甩板弃去孔中液体,沥干水份,用滤纸吸净残留液,然后加入100μl二甲基亚砜,在水平震荡器上反应7-8min,至蓝紫色结晶完全溶解,酶标仪读值,测定在吸收波长为570nm下的od值,记录结果。
实施例5:蛋白免疫印迹(westernblot)实验
1、细胞培养及加药:
(1)取对数生长期hcc827细胞,以胰酶消化后,用含10%胎牛血清的rpmi-1640培养基制成密度为300000个/ml的单细胞悬液,以每孔加入2ml细胞悬液接种至6孔细胞培养板中。
(2)37℃、5%co2培养箱孵育,待细胞贴壁后,实验组加入含不同浓度的药物,1h后加入浓度为1mg/ml白介素-6(il-6)30μl刺激细胞,白介素-6(il-6)终浓度为30ng/ml。
(3)继续培养0.5h后,用ripa裂解液裂解细胞收集蛋白。
2、细胞收集和裂解
(1)去除上层培养基,六孔板中细胞用磷酸盐缓冲液(pbs)洗两次。加入预冷的ripa细胞裂解液(蛋白酶抑制剂和苯甲基磺酰氟与裂解液均以1∶100的比例提前加入混匀)160μl。用提前洗净的细胞刮子将细胞裂解液刮下来,收集到一个干净的1.5ml离心管中。
(2)在冰上放置,裂解30min,每隔一定的时间(6min)涡旋一次。
(3)4℃,12000rpm,离心12min。
(4)将细胞上清液移入到一个干净的离心管中。细胞上清分两部分:取5μl加入到1.5ml的离心管中用于bca测蛋白含量,再加入45μl的1×磷酸盐缓冲液(pbs)混匀备用;剩余的细胞上清定量取140μl,加入5×sds上样缓冲液(loadingbuffer)35μl,混匀后在沸水中煮沸8min,离心后-20℃冰箱中保存。
(5)蛋白浓度测定步骤:
a、1×磷酸盐缓冲液(pbs)稀释蛋白标准品:
b、bca工作液配制:根据标准品和待测样品的个数,计算出总共所需a与b混合工作液的量。按bca试剂a与b体积比50∶1的比例,配制好工作液,涡旋振荡混匀备用。
c、将蛋白标准液和用磷酸盐缓冲液(pbs)稀释好的样品上清液(10倍稀释)各取25μl加入到新的96孔板中。然后再分别加入200μl的提前配好的bca工作液充分混匀。切记不要吹打产生气泡,盖紧96孔板板盖,在37℃恒温箱中反应30min。
d、取出96孔板恢复至室温3-5min,在酶标仪上测定在562nm波长下的吸光度值,并作出标准曲线计算出每个样品的1μl/protein含量以备蛋白上样。
3、十二烷基磺酸钠-聚丙烯酰胺凝胶(sds-page)
(1)固定制胶板,配制10%的sds-page分离胶。
按照下表配制分离胶:10ml
(2)将混好的分离胶分别加入到2块胶板中,加到离顶部1.0cm的位置,用无水乙醇填满胶板,静置30-45min。
(3)分离胶凝好后,倒出剩余的无水乙醇,并用滤纸将剩余无水乙醇吸干净。
(4)按照下表配制5%浓缩胶5ml
(5)将配制好的浓缩胶缓慢地加入胶板中,避免产生气泡,插入样品梳,静置30-45min。
(6)取出蛋白样品,100℃水浴加热5min,转速10000rpm,离心10min。
(7)将胶板固定到电泳槽中,加入sds-page电泳缓冲液,拔出样品梳,按照顺序将处理好的蛋白样品加入到样品槽中。
(8)80v电泳40min。
(9)换电压120v电泳大约1.5h直至溴酚蓝跑出胶体;
4、western-blot转膜
(1)将电泳完毕的sds-page胶放入tbst缓冲液中漂洗一次,蛋白胶放到转膜缓冲液中浸泡。
(2)将一层海绵垫在膜转移缓冲液中浸湿,用镊子夹到转膜仪上,按照海绵垫,三层滤纸,蛋白胶,聚偏氟乙烯(pvdf)膜,三层滤纸,海绵垫,顺序放好,对齐,夹起放到转膜仪上,操作时,滤纸、海绵垫均要在转膜缓冲液中浸湿。每层之间若有气泡应用玻璃试管轻轻滚动将其赶出。
(3)打开转膜仪,300ma转膜75min。
(4)将膜取出,放入tbst缓冲液中,60rpm水平震荡仪漂洗3次,每次8min。
(5)用5%牛血清白蛋白(bsa)封闭液20ml,60rpm水平震荡仪室温封闭2h。
(6)用加有3μl一抗(stat3和p-stat31∶1000)的3ml抗体孵育液,4℃60rpm水平震荡仪过夜孵育。
(7)用10mltbst,常温60rpm水平震荡仪洗涤pvdf膜三次,每次10min。
(8)用加有2μl二抗的20ml抗体孵育液,常温60rpm水平震荡仪孵育pvdf膜2h。
(9)用10mltbst,常温60rpm水平震荡仪洗涤pvdf膜三次,每次10min。
(10)取化学发光底物试剂溶液a和溶液b各1ml,室温显色5min。
(11)用滤纸将膜上的液体吸干,显影仪显影。
实施例6:反转录聚合酶链式反应试验(rt-pcr)
1、细胞培养及加药:
(1)取对数生长期hcc827细胞,通过胰酶消化后,用含10%胎牛血清的rpmi-1640培养基制成密度为100000个/ml的单细胞悬液,以每孔加入2ml细胞悬液接种至6孔细胞培养板中。
(2)37℃、5%co2培养箱孵育,待细胞贴壁后,更换新鲜培养基,实验组加入含不同浓度的药物3,10,30,100(μmol/l),1h后加入浓度为1mg/ml白介素-6(il-6)30μl刺激细胞,白介素-6(il-6)终浓度为30ng/ml。
(3)继续培养24h后,按照rna提取试剂盒说明提取各组细胞的总rna。
2、rna样品制备:
(1)6孔板细胞生长密度为90-100%时,从无菌室取出,弃去上清,每孔加trizol总rna抽提试剂1ml,摇匀,冰上消化5min,使用移液枪头吹打,吸取各孔液体到1.5ml离心管中。
(2)加0.2ml氯仿,轻摇15s,室温静置3min。
(3)4℃,12000rpm离心,15min。离心后液体分为三层,上层为无色水样层rna,中间白色为dna,底层红色为蛋白质,小心吸取上层无色液体转移至一个新的离心管中。
(4)取上清约0.4ml加入等体积异丙醇,室温静置10min。
(5)4℃,12000rpm,离心10min。
(6)弃去离心管中液体,加入75%乙醇1ml。
(7)4℃,7500rpm,离心5min,小心弃去上清,放在超净台中,鼓风干燥5-10min。
(8)加入20-30μl焦炭酸二乙酯(depc)水溶液,分装保存于-70℃冰箱备用。
(9)取2μl的总rna用k5500超微量分光光度计检测浓度与纯度。根据od260/od280的比值鉴定总rna纯度。od260/od280介于1.8-2.0之间说明纯度较好。
(10)取5μl的总rna与上样缓冲液1μl混合,经1%琼脂糖凝胶(含glered),120v,电泳30min(电泳缓冲液为1×tae),凝胶成像分析系统下观察rna的质量。如果rna无降解可行下一步实验。
3、cdna的合成
逆转录聚合酶链反应(reversetranscription-polymerasechainreaction,rt-pcr),实验原理为:首先提取各组细胞中的总rna,以其中的mrna作为模板,在逆转录酶的催化作用下,在随机引物或多聚胸腺嘧啶(oligo(dt))或基因特异性引物的引导下合成互补的dna单链(complementarydna)cdna。再以此cdna为模板,在特异引物的引导下,进行目的基因的pcr扩增反应。由于该cdna是以mrna为模板逆转录得到的,所以扩增的目的基因序列为不包含内含子的可编码基因序列。
(1)cdna的合成
a.取无核酸酶的离心管在冰浴下依次加入以下溶液:
脱氧核糖核苷三磷酸混合物(dntpmix)2.5mm/each4μl;引物混合物(primermix)2ul;rna模板(rnatemplate)1μg;无rna酶水(rnase-freewater)加至15μl,反应条件:70℃孵育10min,迅速冰浴2min。
b.在冰浴中依次加入以下组份,轻轻用移液器混匀:
5倍第一链缓冲液(5×first-strandbuffer)4μl;逆转录酶(m-mlv)1μl,反应条件:42℃温浴50min,85℃加热5min,终止反应。
c.取2-3μl直接用于pcr反应或保存于-20℃。
(2)半定量pcr扩增目的基因
a.在冰上按顺序在pcr管里加入以下反应体系:
cdna2.5μl;正向引物(forwardprimer)(10mm)1μl;反向引物(reverseprimer)(10mm)1μl;2倍pcr预混酶(2×taqpcrmix)12.5ul;双蒸水(ddh2o)补至25μl。
b.pcr反应循环条件:
按以下温度循环:
1)预变性94℃5min;2)变性94℃30s;3)退火61℃30s;4)延伸72℃1min;5)重复2-4步骤30个循环;6)延伸72℃5min;7)4℃保存。
c.结果检测:反应结束后取5μl反应产物,进行琼脂糖凝胶电泳分离鉴定。
- 还没有人留言评论。精彩留言会获得点赞!