一种基于R软件的微生物组差异显著性分析及作图方法与流程
本发明涉及生物信息学领域,具体涉及一种基于r软件的微生物组差异显著性分析及作图方法。
背景技术:
显著性检验就是事先对总体的参数或总体分布形式作出一个假设,然后利用样本信息来判断这个假设是否合理,即判断总体的真实情况与原假设是否存在显著差异。
r是一种用于统计计算和绘图的语言和环境。r提供各种统计(线性和非线性建模,经典统计测试,时间序列分析,分类,聚类......)和绘图的方法,并且具有高度可扩展性。
r的优势之一是可以轻松制作出精心设计的出版品质图片,包括数学符号和公式。它可以在各种unix平台和类似系统(包括freebsd和linux),windows和macos上编译和运行。
从技术上讲,r是一种语法非常简单的表达式语言。基本命令由表达式或赋值组成。如果表达式作为命令给出,则对其进行评估,打印,否则该值将丢失。赋值还会计算表达式并将值传递给变量,但不会自动打印结果。
而现有的分析方法所存在的缺陷是:
(1)结果展示单一:现有的很多基于r的绘图工具,不支持在绘图中添加显著性注释,无法进行相应的展示。
(2)测试方法单一:现有的整合了统计、绘图的工具只提供了单一的测试方法,忽视了样本分布及样本量的影响。
(3)没有考虑多重比较:目前分析流程在判断多组间显著性差异时没有进行多重比较,不能具体说明哪几组均值之间有显著差异。
技术实现要素:
为了克服现有技术所存在的上述缺陷,本发明的目的在于提供一种基于r软件的微生物组差异显著性分析及作图方法。
为了实现本发明的目的之一,所采用的技术方案是:
一种基于r软件的微生物组差异显著性分析及作图方法,包括如下步骤:
数据输入步骤:接收用户输入的测试数据;
测试步骤:根据需要,选择测试方法,所述的测试方法包括方差分析(anova)、t检验、kruskal-wallis检验、wilcoxon检验(非参数方法);其中,所述的方差分析(anova)、t检验是针对正态假设的稳健检验;所述的kruskal-wallis检验、wilcoxon检验(非参数方法)是当违反方差同质性假设时使用;所述的t检验、wilcoxon检验用于比较两个群体的平均值;所述的方差分析、kruskal-wallis检验用于比较两个以上群体间的平均值;
其中方差检验通过stats包aov函数执行,随后通过stats包tukeyhsd函数执行两两比较;
t检验通过stats包t.test函数执行;
kruskal-wallis检验通过stats包kruskal.test函数执行,随后通过pmcmr包posthoc.kruskal.conover.test函数执行两两比较;
wilcoxon检验通过stats包wilcox.test函数执行;
绘图步骤:输出文本测试结果,根据测试数据输出图片;
添加注释步骤:对测试结果结果中不符合假设(平均值不同)的变量进行标注。
在本发明的一个优选实施例中,所述绘图步骤具体是:通过ggplot包geom_boxplot函数绘制箱线图。
在本发明的一个优选实施例中,所述添加注释步骤具体是:对不符合假设的两两群体通过ggsignif包geom_signif函数进行注释,来优化可视化结果。
在本发明的一个优选实施例中,所述的t检验的执行具体为:对若干不同分组样品的测序结果中若干不同物种的丰度的组间差异进行检验。将物种丰度信息按上述要求保存为input.txt文件,分组信息保存为map.txt。在linux终端或者windows命令提示符执行:rscriptcompare_box.rweltch-tinput.txtmap.txt脚本通过for函数,对input.txt中的所有变量在map.txt中所有分组间两两组合的情况进行遍历,并执行weltcht检验,同时保存检验结果;每个变量遍历结束后,通过p.adjust函数对检验结果进行fdr校正,保存校正结果。循环结束后将检验结果以文本形式输出到文件;随后遍历所有输入数据的检验结果,对每一个变量绘制箱线图,并对存在显著差异的分组对进行标注。最后将图形输出到文件中。
在本发明的一个优选实施例中,所述的kruskal-wallis检验的执行具体为:在linux终端或者windows命令提示符执行:rscriptcompare_box.rkruskalinput.txtmap.txt脚本通过for函数,对input.txt中的所有变量进行遍历,并执行kruskal-wallis检验,如果检验结果为存在显著差异,则对所有两两分组执行posthoc检验,并对检验结果进行fdr校正,保存检验结果;循环结束后将检验结果以文本形式输出到文件;随后绘制图形,方法步骤同welcht检验,根据posthoc检验结果进行标注。
本发明的主要创新点在于:
结果信息全面:本方法分析结果中,会在输出图片中添加注释来指示有差异的分组,结果展示更明了易懂。
测试方法全面:本工具整合了anova,kruskal-wallistest,mann-whitney-wilcoxontest,welch’st-test等测试方法,根据不同情况选择使用。
判断更加准确:当统计分析涉及多个同时进行的统计检验时,做出的推论越多,发生错误推论的可能性就越大。通过参数/非参数多重比较方法避免此类错误。
附图说明
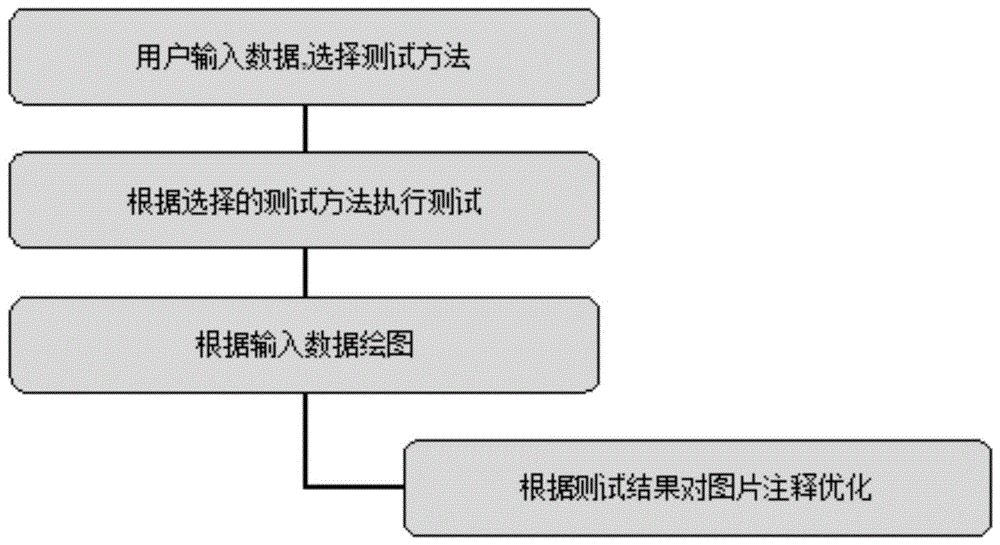
图1为本发明的流程示意图。
图2为stamp软件输出图形示意图。
图3为本发明输出图形示意图。
具体实施方式
以下通过实施例对本发明作进一步的说明,但这些实施例不得用于解释对本发明的限制。
参加图1,本发明实施例所采用的原理是:
1.用户输入用于测试的数据,选择测试方法。
2.对步骤1输入数据执行测试:
方差检验通过stats包aov函数执行,随后通过stats包tukeyhsd函数执行两两比较;
t检验通过stats包t.test函数执行;
kruskal-wallis检验通过stats包kruskal.test函数执行,随后通过pmcmr包posthoc.kruskal.conover.test函数执行两两比较;
wilcoxon检验通过stats包wilcox.test函数执行。
3.对步骤1输入数据进行可视化,通过ggplot包geom_boxplot函数绘制箱线图。
4.根据步骤2测试结果,对不符合假设的两两群体通过ggsignif包
geom_signif函数进行注释。优化可视化结果。
实施例所采用的条件如下:
软件依赖:
r(≧3.1)
pmcmr
ggsignif
ggplot
输入数据:
输入数据要求为制表符分隔文件,第一列为行名,第一行为列名,每列作为一个变量,每行作为一个样本;分组表格式同qiimemapping文件,包含每个样的分组信息。
执行分析:
1.以welcht检验为例,对若干不同分组样品的测序结果中若干不同物种的丰度的组间差异进行检验。将物种丰度信息按上述要求保存为input.txt文件,分组信息保存为map.txt。在linux终端或者windows命令提示符执行:rscriptcompare_box.rweltch-tinput.txtmap.txt脚本通过for函数,对input.txt中的所有变量在map.txt中所有分组间两两组合的情况进行遍历,并执行weltcht检验,同时保存检验结果。
每个变量遍历结束后,通过p.adjust函数对检验结果进行fdr校正,保存校正结果。循环结束后将检验结果以文本形式输出到文件。
随后遍历所有输入数据的检验结果,对每一个变量绘制箱线图,并对存在显著差异的分组对进行标注。最后将图形输出到文件中。
2.以kruskal-wallis检验为例,在linux终端或者windows命令提示符执行:rscriptcompare_box.rkruskalinput.txtmap.txt脚本通过for函数,对input.txt中的所有变量进行遍历,并执行kruskal-wallis检验,如果检验结果为存在显著差异,则对所有两两分组执行posthoc检验,并对检验结果进行fdr校正,保存检验结果。
循环结束后将检验结果以文本形式输出到文件。
随后绘制图形,方法步骤同welcht检验,根据posthoc检验结果进行标注。
参加图2和图3,与图形界面统计软件相比,本发明的方法操作简单,输出图形更加直观,一次性输出全部结果:以stamp软件为例,stamp在执行t检验时无法一次输出全部两两比较结果,需要手动选择需要比较的分组,同时也无法一次输出所有变量的图形结果,在有大量需要比较的变量,多重分组的情况下,类似stamp的鼠标操作的图形界面分析软件需要消耗相当的人力和时间。
同时stamp输出的图形也无法直观的展示检验结果。stamp输出图形见图2,本发明输出图形见图3。
- 还没有人留言评论。精彩留言会获得点赞!