基于增强相互作用网络的关键蛋白质识别方法与流程
本发明属于生物信息学识别关键蛋白质领域,具体地说,是一种通过增强相互作用网络的可靠性、拓扑特征和生物特征来识别关键蛋白质的方法。
背景技术:
蛋白质在细胞的组成和生物体的生命活动中扮演着极其重要的作用。但不同类型的蛋白质对生物体的重要程度不尽相同,其中那些缺失后会导致生物体病变甚至死亡的蛋白质被称为关键蛋白质,其余的则被称为非关键蛋白质。
研究表明,关键蛋白质的识别对于我们了解细胞的生长调控过程,研究生物进化的相关机制,以及根据关键蛋白质进行药物设计、药物标靶鉴定和疾病治疗等方面具有重要意义。
在生物学领域中,识别关键蛋白质通常是采用生物医学实验的方式进行的,这类方法虽然准确,但是成本高、效率低,无法适用于日益增长的蛋白质数据。随着高通量技术的发展,越来越多的蛋白质相互作用数据被获取,这让我们能够从网络水平上识别关键蛋白质。
目前,基于相互作用网络识别关键蛋白质的方法已成为识别关键蛋白质的重要手段之一。相互作用网络是一种复杂网络,具有复杂网络的拓扑特征:小世界、集群或者模块化和无尺度;同时它也是一种生物信息网络,蕴含着生物体在进化、生命活动和细胞组成等方面的生物信息。这为关键蛋白质的识别研究提供了两种思路:基于相互作用网络拓扑特征的识别方法和基于相互作用网络生物信息的识别方法。
其中前者主要是依据“中心性-致死性”法则(centrality-lethalityrule),该法则指出蛋白质分子在网络中的中心性越高,是关键蛋白质的可能性也越高,因而这类方法主要是通过利用相互作用网络中的度、路径、回路和模块化等拓扑特征来度量蛋白质分子的中心性然后识别出关键蛋白质。常用的有8种具有代表性的方法:度中心性(degreecentrality,dc)、介数中心性(betweennesscentrality,bc)、接近度中心性(closenesscentrality,cc)、子图中心性(subgraphcentrality,sc)、特征向量中心性(eigenvectorcentrality,ec)、信息中心性(informationcentrality,ic),局部平均连通度中心性(localaverageconnectivity-based,lac)和邻域中心性(neighborhoodcentrality,nc)。实验结果表明这类方法是有效的,但是由于仅仅使用了相互作用网络的拓扑特征,忽略了网络本身所蕴含的生物信息,因而它们的识别精度不高。
为弥补上述方法的不足,越来越多的研究人员开始研究基于相互作用网络生物信息的识别方法,即通过引入其它生物信息数据来深入挖掘网络本身蕴含着的生物信息。如pec(minl,zhangh,wangj,etal.anewessentialproteindiscoverymethodbasedontheintegrationofprotein-proteininteractionandgeneexpressiondata[j].bmcsystemsbiology,2012,6(1):15.)、coewc(zhangx,xuj,xiaow.anewmethodforthediscoveryofessentialproteins[j].plosone,2013,8(3):e58763.)等方法将基因表达数据引入到相互作用网络中,它们指出关键蛋白质除了成簇出现外,也具有较高的共表达的特性。hc(renj,wangj,lim,etal.discoveringessentialproteinsbasedonppinetworkandproteincomplex[j].internationaljournalofdatamining&bioinformatics,2015,12(1):24.)、uc(lim,luy,niuz,etal.unitedcomplexcentralityforidentificationofessentialproteinsfromppinetworks[j].ieee/acmtransactionsoncomputationalbiology&bioinformatics,2017,14(2):370-380.)等方法则结合了相互作用网络和蛋白质复合物数据,它们发现参与形成更多复合物的蛋白质更有可能是关键蛋白质。ion算法(pengw,wangj,wangw,etal.iterationmethodforpredictingessentialproteinsbasedonorthologyandprotein-proteininteractionnetworks[j].bmcsystemsbiology,2012,6(1):1-17.)则基于关键蛋白质在生物进化过程中更加保守的特性,通过随机游走模型结合了相互作用网络和蛋白质直系同源的信息来识别关键蛋白质。实验结果表明,这类方法的识别精度普遍优于前者。
上述两类方法在识别精度、识别效率等方面均取得了显著成果,但是依然存在着一些问题:一方面,这些方法对相互作用网络本身可靠性的依赖程度较高,而当前蛋白质相互作用数据库中所收集的相互作用信息通常包含了大量的假阴性和假阳性数据,这些数据会降低网络的可靠性,进而影响这些方法的识别精度;另一方面,研究表明,蛋白质的关键性主要体现在其所承担功能的特有性上,这种功能特有性通常意味着蛋白质结构组成上的特有性,而现有的方法对蛋白质的这种结构组成上的生物特征考察不足。因此,如何降低相互作用网络中假阴性和假阳性数据的影响、挖掘相互作用网络蕴含的生物特征是提高现有方法识别精度的关键。
技术实现要素:
本发明的目的在于提供一种一种基于增强相互作用网络的关键蛋白质识别方法。
实现本发明目的的技术解决方案为:一种基于增强相互作用网络的关键蛋白质识别方法,包括以下步骤:
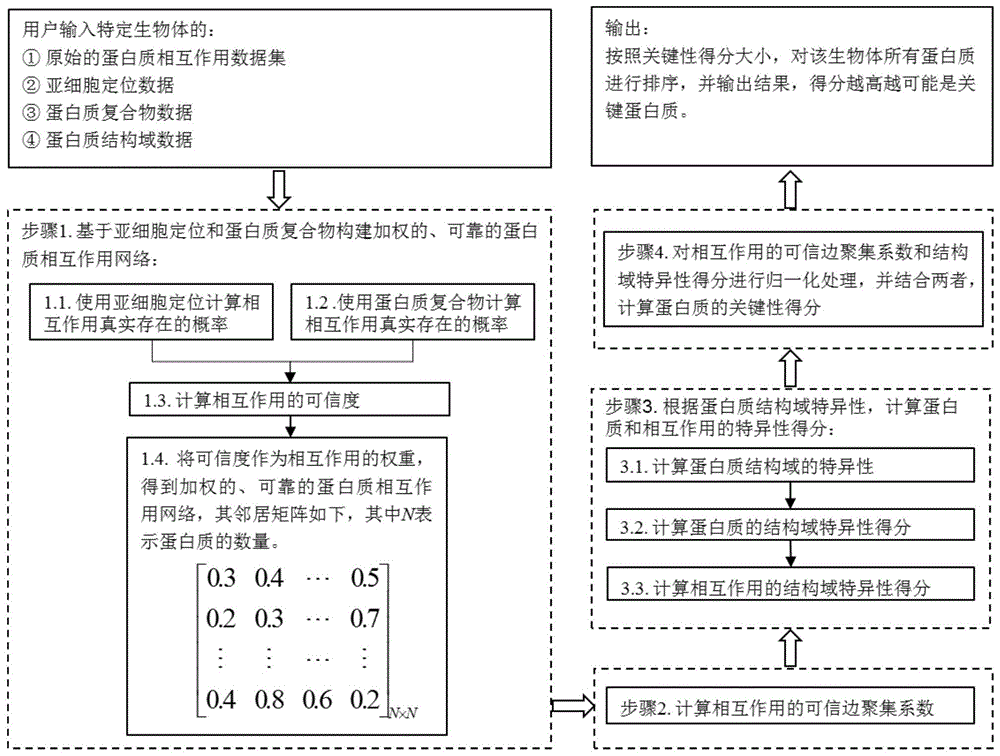
步骤1:基于亚细胞定位、蛋白质复合物构建加权的、可靠的蛋白质相互作用网络。针对原始蛋白质相互作用网络中的任一条相互作用,首先统计其对应的两个蛋白质分子出现的相同亚细胞定位的数量,数量越多表明这条相互作用真实存在的可能性越高,将相同亚细胞定位的数量归一化处理后表示为这条相互作用真实存在的概率;其次统计这条相互作用对应的两个蛋白质分子参与形成的相同蛋白质复合物的数量,数量越多表明这条相互作用真实存在的可能性越高,将相同蛋白质复合物的数量归一化处理后表示为这条相互作用真实存在的概率;接着,结合上述两个概率,计算这条相互作用的可信度;最后将计算出的可信度作为该条相互作用的权重,由此构建出一个可靠的、加权的蛋白质相互作用网络。
步骤2:计算网络中每条相互作用的可信边聚集系数。针对步骤1中构建的加权的蛋白质相互作用网络,使用可信边聚集系数计算每条相互作用在网络中所处位置的集聚程度。
步骤3:基于蛋白质结构域特异性,计算网络中蛋白质分子和相互作用的结构域特异性得分。借鉴tf-idf算法的思想,将蛋白质分子和结构域分别视作文本文件和单词进行处理,进而分析蛋白质的结构特征。首先计算每个结构域的特异性;然后依据结构域在蛋白质中出现的频率和自身的特异性,计算出每个蛋白质的结构域特异性得分;最后,针对任一条相互作用,将其对应的两个蛋白质分子的结构域特异性得分取平均,从而得到这条相互作用的结构域特异性得分。
步骤4:对上述步骤计算出的相互作用的可信边聚集系数和结构域特异性得分进行归一化处理,结合两者,计算每个蛋白质分子的关键性得分,根据得分高低对蛋白质分子进行排序并输出结果。
本发明与现有技术相比,其显著优点:(1)通过亚细胞定位和蛋白质复合物对相互作用的可信度进行评估,可以降低相互作用网络中假阴性和假阳性数据的影响,增强网络的可靠性;(2)使用可信边聚集系数计算相互作用在加权网络中所处位置的集群程度,不仅能够体现出相互作用可信度的意义,也更符合关键蛋白质更可能和关键蛋白质产生相互作用且成簇出现的特性,因而可以有效增强相互作用网络的拓扑特征;(3)基于蛋白质结构域特异性,可以得到蛋白质在结构组成上特有性的特征,从而发现并增强相互作用网络蕴含的生物特征;(4)可信边聚集系数和结构域特异性得分分别能够有效描述相互作用网络的拓扑信息和生物信息,通过结合两者,可以更加准确且全面地评估蛋白质的关键性,提高方法的识别精度。
附图说明
图1为基于增强相互作用网络的关键蛋白质识别方法流程结构示意图。
图2为酵母的蛋白质相互作用网络。
图3为蛋白质的结构域组成示意图。
具体实施方式
下面结合说明书附图和实施例对本发明作进一步说明。
图1给出了本发明的流程结构示意图。结合图1所示,本发明一种基于增强相互作用网络的关键蛋白质识别方法,包括以下步骤:首先,为增强网络的可靠性,使用亚细胞定位和蛋白质复合物来度量原始的相互作用网络中每条相互作用真实存在的可能性即可信度,并将其作为每条相互作用的权重,从而构建起一个加权的、可靠的相互作用网络;其次,为增强网络的拓扑特征,针对加权网络中的每条相互作用,使用可信边聚集系数计算其在网络中所处位置的集聚程度;接着,为增强网络的生物特征,根据蛋白质结构域特异性,计算加权网络中每个蛋白质分子的结构域特异性得分,并进一步计算出每条相互作用的结构域特异性得分;最后,结合相互作用的可信边聚集系数和结构域特异性得分,计算网络中每个蛋白质分子的关键性得分,并根据得分高低对蛋白质分子进行排序,并输出结果。
考虑到酵母的蛋白质相互作用数据的相对完备性,实施例选择它作为研究对象。其中酵母的相互作用数据是从dip数据库下载获得,去除冗余和自连接的相互作用后,共提取出22620条相互作用,包含5126个蛋白质分子。酵母的亚细胞定位数据来源于compartment数据库,经过预处理后,共获得mitochondrion、nucleus等11种亚细胞定位,以及35223条蛋白质的亚细胞定位分布信息。酵母的蛋白质复合物数据是通过整合cm270、cm425、cyc408和cyc428四种已知的蛋白质复合物数据集得到,去除冗余数据后,共获得745个蛋白质复合物信息,与从dip数据库中获取的酵母的5126个蛋白质分子匹配后,共提取2167个蛋白质分子参与形成的蛋白质复合物信息。酵母的蛋白质结构域数据是从pfam数据库中下载获得,采用的数据集版本是于2017年3月份更新的pfam31.0。另外,在pfam数据库中,有两种不同质量水平的结构域序列数据:pfam-a系列和pfam-b系列,由于pfam-b系列的数据未经注释且质量水平较低,本实施例仅获取酵母的pfam-a系列的结构域序列数据并用于实验,通过与从dip数据库中获取的酵母的5126个蛋白质分子匹配后,共提取出具有已知的结构域信息的蛋白质共有4174个,包含了2829种结构域。
下面将结合附图所示,更加具体地描述前述过程。
步骤1:基于亚细胞定位、蛋白质复合物构建加权的、可靠的相互作用网络从dip数据库中获取到酵母的蛋白质相互作用数据集,可以表示为一个无权的复杂网络,即将蛋白质分子表示成网络中的节点,相互作用表示成网络中的边,如图2所示。但是由原始数据集构建出的网络通常包含了大量的假阴性和假阴性数据,为增强网络的可靠性,本实施例使用亚细胞定位和蛋白质复合物计算该网络中每条相互作用真实存在的可能性即可信度,并将计算出的可信度作为相互作用的权重,由此构建出一个加权的、可靠的相互作用网络。具体构建方式按照如下步骤执行:
步骤1.1:使用亚细胞定位计算相互作用真实存在的概率。针对酵母的任一条相互作用e(u,v),其对应的两个蛋白质分子为u和v,统计u和v在上述获取的酵母的亚细胞定位分布信息中共同出现的亚细胞定位的数量,表示为subcellular(u,v),则相互作用e(u,v)真实存在的概率为:
其中,subcellularmax表示酵母的所有相互作用对应的两个蛋白质共同出现的亚细胞定位最多的数量。分子、分母同时加1,是为了保证蛋白质u和v没有共同出现在任一亚细胞定位的时候,它们之间相互作用真实存在的概率不等于0。
步骤1.2:使用蛋白质复合物计算相互作用真实存在的概率。针对酵母的任一条相互作用e(u,v),在上述获取的酵母的蛋白质复合物数据集中,统计其对应的两个蛋白质u和v参与形成相同复合物的数量,表示为complex(u,v),则相互作用e(u,v)真实存在的概率为:
其中,complexmax表示酵母的所有相互作用对应的两个蛋白质参与形成相同蛋白质复合物最多的数量。
步骤1.3:计算相互作用的可信度。通过结合步骤1.1和步骤1.2,对于酵母的任一条相互作用e(u,v),其可信度用p-subcellular(u,v)和p-complex(u,v)表示为:
步骤1.4:构建加权的、可靠的相互作用网络。将步骤1.3计算的可信度作为网络中对应相互作用的权重,从而将酵母的原始的无权的网络变成一个加权的网络,且由于考虑了每条相互作用真实存在的可能性,因而更加值得信赖,本实施例将构建后的网络表示为re-pin(reliableproteininteractionnetwork),令a=wu,v表示其邻接矩阵,则a=wu,v是一个5126×5126形式的矩阵,且如果蛋白质u和v之间存在一条相互作用,则wu,v=bel(u,v)∈(0,1],表示这条相互作用的可信度,否则wu,v=0,如下:
步骤2:计算re-pin中相互作用的可信边聚集系数。
对于网络中的任一条边,边聚集系数被定义为该边在网络中实际参与构成的三角形个数与该边最多可能参与构成的三角形个数之比。但是由于边聚集系数不适用于加权的网络,不能体现权重的意义,本实施例使用改进后的可信边聚集系数(reliableedgeclusteringcoefficient,re-ecc)计算相互作用在网络中所处位置的集聚程度。对于re-pin中任一条相互作用e(u,v),其可信边聚集系数表示为:
其中,nw表示蛋白质u和v相同的邻居节点的集合,即在相互作用网络re-pin中与蛋白质节点u和v能够形成三角形的节点的集合。triangle(u,v,w)表示由蛋白质节点u、v和w形成三角形的概率,其计算方式主要基于re-pin中相互作用的权重,即步骤1.3计算出的每条相互作用的可信度,如下:
triangle(u,v,w)=bel(u,v)×bel(u,w)×bel(v,w)(19)
degree(u)和degree(u)分别表示蛋白质节点u和v在加权网络re-pin中的度数,以degree(u)为例,其计算方式如下:
其中,ns表示蛋白质节点u的邻居节点的集合,s是u的邻居节点且s∈ns。
步骤3:基于蛋白质结构域特异性计算蛋白质和相互作用的结构域特异性得分。
蛋白质在结构组成上的特有性是评估蛋白质关键性的有效指标,本实施例使用酵母的蛋白质结构域信息分析其蛋白质的结构特征。借鉴tf-idf算法的思想,将每种类型的蛋白质结构域视作一个单词,那么每条蛋白质就相当于一个文本文件,则酵母包含的所有蛋白质就相当于一个语料库。如将pf00270、pf00271等结构域视作一种单词,则蛋白质yer172c、ybl084c、ydl126c、ydl145c的“文本”组成可以表示如图3所示。本实施例根据蛋白质结构域的特异性,计算re-pin中每个蛋白质的结构域特异性得分,并进一步计算出每条相互作用的结构域特异性得分。具体计算方式按照如下步骤执行:
步骤3.1:使用ipf(inverseproteinfrequency)计算结构域的特异性。对于酵母的任一结构域d,先计算酵母包含的蛋白质总数除以包含该结构域的蛋白质数目加1(避免分母等于0的情况),再将得到的商数取对数,得出其特异性,如下:
其中,|p(s)|表示酵母包含的蛋白质总数5126,|{p:d∈p}|表示包含结构域d的蛋白质的数目。
步骤3.2:使用df(domainfrequency)计算结构域在蛋白质中出现的频率。对于酵母的任一结构域d,其在蛋白质p中出现的频率,表示为这个结构域在蛋白质p中出现的次数和蛋白质p包含的结构域的总数之比,如下:
其中,nd,p表示蛋白质结构域d在蛋白质分子p中出现的频次,k表示蛋白质分子p包含的结构域种类数,则∑knk,p表示蛋白质分子p包含的结构域的总数。
步骤3.3:计算蛋白质的结构域特异性得分。结合步骤3.1和步骤3.2,对于酵母的任一蛋白质p,计算其包含的每种类型结构域的特异性与该类型结构域在此蛋白质p中出现的频率乘积之和,得出其结构域特异性得分,如下:
specificity(p)=∑kdfd,p×ipfd(23)
其中,表示k蛋白质p包含的结构域的种类。
步骤3.4:计算相互作用的结构域特异性得分。对于re-pin中任一条相互作用e(u,v),通过计算其对应的两个蛋白质u和v的结构域特异性得分的平均值,得到这条相互作用的结构域特异性得分,如下:
步骤4:融合可信边聚集系数和结构域特异性得分,计算蛋白质的关键性得分
可信边聚集系数和结构域特异性得分能够有效描述相互作用网络的拓扑信息和生物信息,本实例通过结合两者,更加准确且全面地评估蛋白质的关键性。对于任一条相互作用e(u,v),先对其在步骤2和步骤3中得到的可信边聚集系数re-ecc(u,v)和结构域特异性得分specificity(u,v)进行归一化处理,如下:
其中,re-eccmax和re-eccmin分别表示re-pin中所有相互作用的可信边聚集系数的最大值和最小值;specificitymax和specificitymin分别表示re-pin中所有相互作用的结构域特异性得分的最大值和最小值。
则对于酵母的任一蛋白质u,其关键性得分可以表示为
其中nv是蛋白质u的邻居节点的集合,v是节点的邻居节点且v∈nv。计算出酵母的所有蛋白质的关键性得分后,根据得分高低排序,得分越高越可能是关键蛋白质,并输出结果。
本发明使用亚细胞定位和蛋白质复合物来度量原始的相互作用网络中相互作用真实存在的可能性,并将其作为相互作用的权重,从而构建起一个加权的、可靠的网络;使用蛋白质结构域分析蛋白质在结构组成上的特征。这样处理能够有效降低网络中假阴性和假阳性数据的影响,增强网络的可靠性,并获取到相互作用网络蕴含的生物特征。另外,由于现有的网络拓扑特征如节点的度数、路径、子图等不适用于识别关键蛋白质的研究,因而本发明使用可信边聚集系数来刻画加权相互作用网络的拓扑特征。综上所述,本发明通过增强相互作用网络的可靠性、拓扑特征和生物特征,来提高关键蛋白质的识别精度。
- 还没有人留言评论。精彩留言会获得点赞!