一种基于差分进化的蛋白质ATP对接方法与流程
本发明涉及一种生物信息学、智能优化、计算机应用领域,尤其涉及的是,一种基于差分进化的蛋白质atp对接方法。
背景技术:
随着对蛋白质的不断深入研究发现,蛋白质与小分子或配体结合的现象普遍存在,尤其是蛋白质与能量分子的结合更是广泛存在于各种生命现象中,因此研究蛋白质与配体结合的特性和规律是十分必要的。atp是一种不稳定的高能化合物,又称腺苷三磷酸。水解时释放出能量较多,是生物体内最直接的能量来源。在细胞中,它能与adp相互转化实现贮能和放能,从而保证了细胞各项生命活动的能量供应。生物体内很多重要的生理过程,如细胞的周期调控、合成代谢、信号传导以及遗传信息的传递等,都依赖于蛋白质和配体分子的相互作用和识别。分子对接方法对生命活动的分子机理研究、生物分子复合物结构预测和靶向药物筛选等都具有重要的意义。
经典的热力学认为,蛋白质和配体分子相互作用所形成的复合物结构应该是结合自由能最低的构象,快速且准确搜索能量极低的构象对于蛋白质-配体分子对接至关重要。
因此,分子对接计算需要采用数学模型或函数尽量准确的计算结合自由能,而且还需要高效的搜索算法快速找到自由能极低的构象。分子对接中的构象搜索是极其复杂的问题,蛋白质-配体分子对接一方面需要搜索能量低的构象,另一方面需要在短时间内搜索到各种可能的情况,因此快速有效的搜索算法是分子对接中重要的研究领域。蛋白质-配体分子对接构象搜索方法主要包括快速穷举搜索和启发式搜索两大类。配体相互作用的区域可能出现在分子表面任何位置,因此往往需要进行全局搜索,可采用快速穷举搜索遍历各种位置,或采用启发式算法进行近似的全局搜索。
快速穷举算法虽然可以快速搜索整个构象空间,但同时也引入了较多错误的构象,为区分正确构象增加了难度。启发式搜索算法是把对接体系中配体分子先进行随机平移和旋转操作,并根据能量评分对操作后的配体构象进行优化和取舍,最终找到能量最低的配体分子构象。而启发式的蒙特卡洛算法是一种通用的搜索方法,它可以在配体构象空间中随机采样,且不受构象空间结构和分布的影响。但该方法可能需要较长的计算时间才能给出较优的解。rosettadock程序(wangc,schueler-furmano,bakerd.improvedside-chainmodelingforprotein-proteindocking[j].proteinscience,2005,14(5):1328-1339.)和glide程序(halgrenta,murphyrb,friesnerra,etal.glide:anewapproachforrapid,accuratedockingandscoring.2.enrichmentfactorsindatabasescreening[j].journalofmedicinalchemistry,2004,47(7):1750-1759.)就是采用蒙特卡洛算法进行全空间构象搜索,而rosettadock程序的分子对接过程往往需要较长的时间。
因此,现有的蛋白质atp分子对接方法在计算代价和搜索效率方面存在着缺陷,需要改进。
技术实现要素:
为了克服现有的蛋白质与atp对接方法在计算代价、预测精确性方面的不足,本发明提出一种计算代价低、预测精确性高的基于差分进化算法的蛋白质atp对接方法。
本发明解决其技术问题所采用的技术方案是:
一种基于差分进化的蛋白质atp对接方法,所述方法包括以下步骤:
1)输入蛋白质和atp的结构信息,分别记为r和a;
2)对于输入的结构信息r,使用atpbind服务器(https://zhanglab.ccmb.med.umich.edu/atpbind/)预测出蛋白质-atp绑定的残基位点信息,得出蛋白质与atp绑定的n个残基,分别记为r1,r2,...,rn;
3)根据r1,r2,...,rn的中心碳原子cα坐标信息聚类出一个中心点cr,根据a中各原子坐标信息聚类出一个中心点ca,移动atp使得ca和cr这两点的坐标重合;
4)根据a中各原子的坐标信息聚类为三个中心点,三个中心点被称为拟原子,分别表示为
5)对pdb数据库中的每个atp分子a(j),j=1,2,...,n,根据它所有原子的坐标信息聚类出与
6)对pdb数据库中每个atp的每个中心点
7)计算任意残基类型t与pdb数据库中所有atp分子的第k个中心原子ck,k=1,2,3,相互作用的平均距离,记作d(ck,t):
其中
8)根据步骤7),分别计算pdb数据库中所有t类型残基与其绑定的atp中心点ck的相互作用平均距离d(ck,t);
9)参数设置:设置种群规模np,缩放因子f,交叉概率cr,最大迭代次数gmax,初始化迭代次数g=0;
10)种群初始化:随机生成初始化种群p={s1,s2,...,si,...,snp},si=(si,1,si,2,si,3,si,4,si,5,si,6)为种群p的第i个个体,si,1、si,2、si,3、si,4、si,5与si,6为si的6个元素,其中si,1、si,2和si,3的取值范围是
11)对于种群中的每个个体si,根据如下方式将蛋白质与atp对接,并计算该个体的得分score(si):
11.1)根据si中的后三个元素si,4、si,5与si,6,计算出一个三维空间旋转矩阵r:
11.2)将
11.3)根据si中的前三个元素si,1、si,2、si,3,将旋转得到的坐标
其中c'k是平移后得到的三维坐标,对于c'k和
11.4)根据步骤8),计算得分score(si):
score(si)=∑|dkt-d(ck,t)|
其中dkt是c'k与残基类型为t的残基cα原子的距离,k=1,2,3;
12)根据差分进化算法,对种群p中的每个个体si,i∈{1,2,…,np}作如下处理:
12.1)从当前种群中p随机选择三个不同的个体sa、sb与sc,其中a≠b≠c≠i,根据如下等式生成一个突变个体smutant:
smutant=sa+f·(sb-sc)
12.2)将si中的元素信息复制到交叉个体scross中,再在scross的6个元素中随机选择一个元素scross,j,使用smutant中对应的元素smutant,j替换,最后,对于scross中的每一个元素,使用随机生成的0到1之间的随机数r来控制是否使用smutant中对应的元素来替换:若r<cr,则替换,否则不替换;
12.3)根据步骤11),分别计算scross与si对应的得分score(scross)与score(si);
12.4)如果score(sscore)<score(si),则使用scross替换种群p中的si,否则si保留在种群p中;
13)g=g+1,如果g>gmax,则根据当前种群p中得分最低的个体slow,将a中所有原子坐标根据slow中的元素信息进行旋转平移后的坐标作为最终的配体位置信息输出,否则返回步骤12)。
本发明的技术构思为:首先,使用atpbind服务器预测出蛋白质atp绑定残基信息,提升了复合物分子空间结构的预测精度;然后,通过种群个体的设计将原先的蛋白质-atp结构预测问题转换成搜索最优个体的优化问题,降低了计算代价;最后,通过使用差分进化算法搜索最优个体,提升了蛋白质-atp复合物结构的预测精度。本发明提供一种计算代价低、搜索效率高的基于差分进化的蛋白质-atp对接方法。
本发明的有益效果表现在:一方面,使用atpbind服务器预测蛋白质-atp绑定残基信息,提升了蛋白质-atp复合物分子空间结构的预测精度;另一方面,将蛋白质-atp对接预测问题转换成选择最优个体的优化问题,并使用差分进化算法搜索最优个体,提高了蛋白质-atp对接预测的效率与准确性。
附图说明
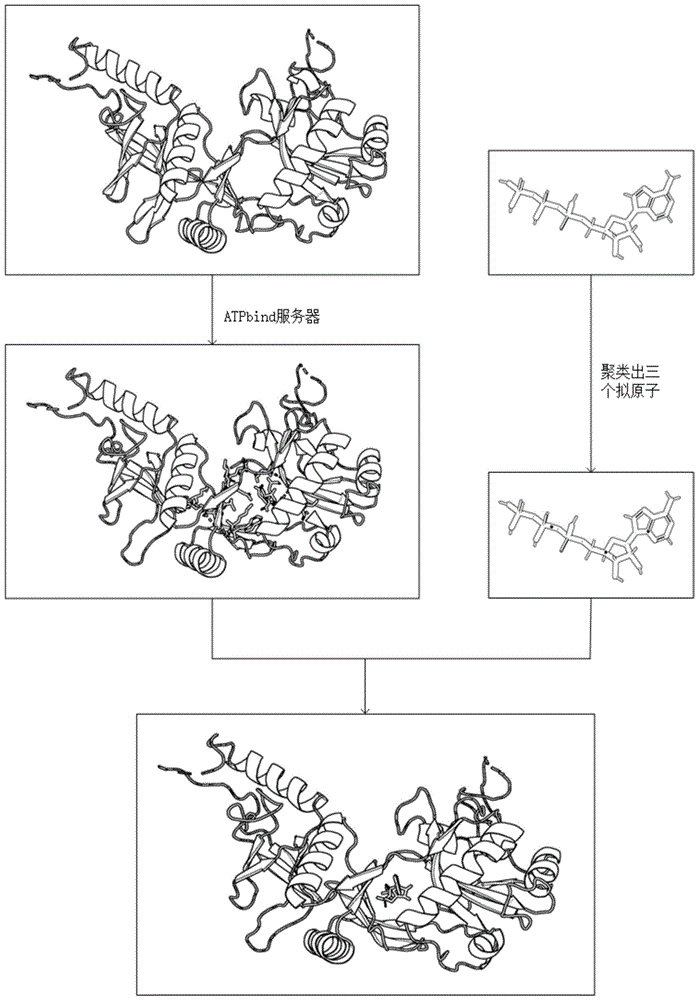
图1为一种基于差分进化的蛋白质atp对接方法的示意图。
图2为使用一种基于差分进化的蛋白质atp对接方法对蛋白质1a0i与atp进行预测后得到复合物三维空间结构图。
具体实施方式
下面结合附图对本发明作进一步描述。
参照图1和图2,一种基于差分进化的蛋白质与atp对接方法,包括以下步骤:
1)输入蛋白质和atp的结构信息,分别记为r和a;
2)对于输入的结构信息r,使用atpbind服务器(https://zhanglab.ccmb.med.umich.edu/atpbind/)预测出蛋白质-atp绑定的残基位点信息,得出蛋白质与atp绑定的n个残基,分别记为r1,r2,...,rn;
3)根据r1,r2,...,rn的中心碳原子cα坐标信息聚类出一个中心点cr,根据a中各原子坐标信息聚类出一个中心点ca,移动atp使得ca和cr这两点的坐标重合;
4)根据a中各原子的坐标信息聚类为三个中心点,三个中心点被称为拟原子,分别表示为
5)对pdb数据库中的每个atp分子a(j),j=1,2,...,n,根据它所有原子的坐标信息聚类出与
6)对pdb数据库中每个atp的每个中心点
7)计算任意残基类型t与pdb数据库中所有atp分子的第k个中心原子ck,k=1,2,3,相互作用的平均距离,记作d(ck,t):
其中
8)根据步骤7),分别计算pdb数据库中所有t类型残基与其绑定的atp中心点ck的相互作用平均距离d(ck,t);
9)参数设置:设置种群规模np,缩放因子f,交叉概率cr,最大迭代次数gmax,初始化迭代次数g=0;
10)种群初始化:随机生成初始化种群p={s1,s2,...,si,...,snp},si=(si,1,si,2,si,3,si,4,si,5,si,6)为种群p的第i个个体,si,1、si,2、si,3、si,4、si,5与si,6为si的6个元素,其中si,1、si,2和si,3的取值范围是
11)对于种群中的每个个体si,根据如下方式将蛋白质与atp对接,并计算该个体的得分score(si):
11.1)根据si中的后三个元素si,4、si,5与si,6,计算出一个三维空间旋转矩阵r:
11.2)将
11.3)根据si中的前三个元素si,1、si,2、si,3,将旋转得到的坐标
其中c'k是平移后得到的三维坐标,对于c'k和
11.4)根据步骤8),计算得分score(si):
score(si)=∑|dkt-d(ck,t)|
其中dkt是c'k与残基类型为t的残基cα原子的距离,k=1,2,3;
12)根据差分进化算法,对种群p中的每个个体si,i∈{1,2,…,np}作如下处理:
12.1)从当前种群中p随机选择三个不同的个体sa、sb与sc,其中a≠b≠c≠i,根据如下等式生成一个突变个体smutant:
smutant=sa+f·(sb-sc)
12.2)将si中的元素信息复制到交叉个体scross中,再在scross的6个元素中随机选择一个元素scross,j,使用smutant中对应的元素smutant,j替换,最后,对于scross中的每一个元素,使用随机生成的0到1之间的随机数r来控制是否使用smutant中对应的元素来替换:若r<cr,则替换,否则不替换;
12.3)根据步骤11),分别计算scross与si对应的得分score(scross)与score(si);
12.4)如果score(sscore)<score(si),则使用scross替换种群p中的si,否则si保留在种群p中;
13)g=g+1,如果g>gmax,则根据当前种群p中得分最低的个体slow,将a中所有原子坐标根据slow中的元素信息进行旋转平移后的坐标作为最终的配体位置信息输出,否则返回步骤12)。
本实施例以预测蛋白质1a0i和atp对接后的复合物的三维空间结构为实施例,一种基于差分进化的蛋白质atp对接方法,包括以下步骤:
1)输入蛋白质和atp的结构信息,分别记为r和a;
2)对于输入的结构信息r,使用atpbind服务器(https://zhanglab.ccmb.med.umich.edu/atpbind/)预测出蛋白质-atp绑定的残基位点信息,得出蛋白质与atp绑定的n个残基,分别记为r1,r2,...,rn;
3)根据r1,r2,...,rn的中心碳原子cα坐标信息聚类出一个中心点cr,根据a中各原子坐标信息聚类出一个中心点ca,移动atp使得ca和cr这两点的坐标重合;
4)根据a中各原子的坐标信息聚类为三个中心点,三个中心点被称为拟原子,分别表示为
5)对pdb数据库中的每个atp分子a(j),j=1,2,...,n,根据它所有原子的坐标信息聚类出与
6)对pdb数据库中每个atp的每个中心点
7)计算任意残基类型t与pdb数据库中所有atp分子的第k个中心原子ck,k=1,2,3,相互作用的平均距离,记作d(ck,t):
其中
8)根据步骤7),分别计算pdb数据库中所有t类型残基与其绑定的atp中心点ck的相互作用平均距离d(ck,t);
9)参数设置:设置种群规模np,缩放因子f,交叉概率cr,最大迭代次数gmax,初始化迭代次数g=0;
10)种群初始化:随机生成初始化种群p={s1,s2,...,si,...,snp},si=(si,1,si,2,si,3,si,4,si,5,si,6)为种群p的第i个个体,si,1、si,2、si,3、si,4、si,5与si,6为si的6个元素,其中si,1、si,2和si,3的取值范围是
11)对于种群中的每个个体si,根据如下方式将蛋白质与atp对接,并计算该个体的得分score(si):
11.1)根据si中的后三个元素si,4、si,5与si,6,计算出一个三维空间旋转矩阵r:
11.2)将
11.3)根据si中的前三个元素si,1、si,2、si,3,将旋转得到的坐标
其中c'k是平移后得到的三维坐标,对于c'k和
11.4)根据步骤8),计算得分score(si):
score(si)=∑|dkt-d(ck,t)|
其中dkt是c'k与残基类型为t的残基cα原子的距离,k=1,2,3;
12)根据差分进化算法,对种群p中的每个个体si,i∈{1,2,…,np}作如下处理:
12.1)从当前种群中p随机选择三个不同的个体sa、sb与sc,其中a≠b≠c≠i,根据如下等式生成一个突变个体smutant:
smutant=sa+f·(sb-sc)
12.2)将si中的元素信息复制到交叉个体scross中,再在scross的6个元素中随机选择一个元素scross,j,使用smutant中对应的元素smutant,j替换,最后,对于scross中的每一个元素,使用随机生成的0到1之间的随机数r来控制是否使用smutant中对应的元素来替换:若r<cr,则替换,否则不替换;
12.3)根据步骤11),分别计算scross与si对应的得分score(scross)与score(si);
12.4)如果score(sscore)<score(si),则使用scross替换种群p中的si,否则si保留在种群p中;
13)g=g+1,如果g>gmax,则根据当前种群p中得分最低的个体slow,将a中所有原子坐标根据slow中的元素信息进行旋转平移后的坐标作为最终的配体位置信息输出,否则返回步骤12)。
以预测蛋白质1a0i和atp对接的三维空间结构为实施例,运用以上方法得到的蛋白质1a0i和atp的复合物三维空间结构如图2所示。
以上说明是本发明以蛋白质1a0i和atp为实例所得出的预测结果,并非限定本发明的实施范围,在不偏离本发明基本内容所涉及范围的前提下对其做各种变形和改进,不应排除在本发明的保护范围之外。
- 还没有人留言评论。精彩留言会获得点赞!