腺苷酸琥珀酸合成酶基因和/或其编码的蛋白的新应用的制作方法
本发明属于生物医药领域,具体涉及腺苷酸琥珀酸合成酶基因和/或其编码的蛋白的新应用。
背景技术:
急性髓系白血病(acutemyeloidleukemia,aml)是一种起源于造血干/祖细胞的恶性克隆性疾病,主要表现为髓系原始和幼稚细胞增殖失控、凋亡受阻、分化停滞,进而抑制正常造血,并引起髓外浸润。aml发病机制复杂、临床预后差,尽管联合化疗使大多数患者可获得完全缓解(completeremission,cr),但只有少数特殊类型的aml患者能够长期存活,大部分患者在cr后的3年内发生疾病复发,且难治或复发患者化疗效果差,因此针对aml发生发展中关键分子为靶点的个体化治疗将成为aml治疗的发展趋势。目前关于aml的机制研究仍不充分,现有的分子靶向药物难以满足临床治疗的需要,因此挖掘和寻找aml发生发展中新的关键致病分子和潜在干预靶点,为诊疗分层和预后评估提供新的依据具有至关重要的临床意义。
技术实现要素:
联合化疗是急性髓系白血病的主要干预策略,但由于化疗的无选择性不可避免地会伤害正常细胞,引起毒副作用,因此针对aml发生发展中关键分子为靶点的精准靶向治疗将成为aml治疗的新趋势。目前肿瘤的分子靶向治疗已取得巨大进步,如酪氨酸激酶抑制剂、cd20单抗等分子靶向药物已显示良好的临床疗效。然而,aml的发生发展是一个多因素、多阶段、多步骤的过程,涉及到大量分子参与的复杂网络调控,现有的分子靶向药物难以满足临床治疗的需要。因此,挖掘aml发生发展新的节点分子,确定其做为aml干预的潜在靶点具有十分重要的价值。此外,目前基于细胞遗传学的aml风险分层仍然不够精确,尽管大多数研究已经在特定的aml亚群中进行,但引入新的遗传和表观遗传标志物有助于缩小这一差距并提高风险分层的特异性。
随着生物技术的发展,尤其是以信息学为手段的分析方法的推陈出新,让人们对于数据的收集、处理、分析及使用有了更大的需求和更深的理解,而利用生物标志物进行诊断和治疗也日益受到关注。同时,现代生物技术的进步可以使得研究者从不同的层面全方位地了解肿瘤的发生、发展,作为一种初步定位标记物,分析、筛选相应的分子靶标,甚至是相关药物筛选、临床应用探究的工具意义重大,为人类寻求多水平的治疗手段提供了依据,人们可以根据癌症患者肿瘤相关基因和分子的变异情况设计出个体化的治疗程序。因此,利用生物信息学的方法快速寻找和定位全新的aml诊断和预后标记物具有重大意义。
腺苷酸琥珀酸合成酶(adss)定位于12号染色体远端,是嘌呤生物合成中的必须酶,利用用鸟苷5'-一磷酸(gtp)和天冬氨酸分子通过几种动力学中间体将肌苷5'-一磷酸(imp)转化为琥珀酰腺苷5'-一磷酸(samp),从而影响腺嘌呤的生物合成。已有报道证实adss的缺失和高尿酸血症以及痛风密切相关,且有研究表明腺苷酸琥珀酸合成酶1(adss1)基因是肺腺癌中新的缺失靶点,与原发性小鼠肿瘤中染色体不稳定表型显著相关。但adss在血液系统疾病中的作用尚不明确,本专利基于对tcga数据库和geo数据库的数据分析发现aml患者相对正常对照组体内adss表达降低,且adss低表达组aml患者具有更差的预后,提示其可能参与aml发生发展。
为解决现有技术上的不足,根据研究结果,本发明提供了腺苷酸琥珀酸合成酶基因和/或其编码的蛋白的新应用。
本发明是通过以下技术方案实现的:
腺苷酸琥珀酸合成酶基因和/或其编码的蛋白在制备急性髓系白血病治疗药物中的应用。
优选的,所述药物中包括腺苷酸琥珀酸合成酶基因和/或其编码的蛋白,以及一种或多种药用赋形剂或药用载体。
腺苷酸琥珀酸合成酶基因和/或其编码的蛋白在制备急性髓系白血病辅助诊断和/或预后判断的组合物中的应用。
优选的,所述的辅助诊断和/或预后判断的组合物包括:用于在pcr中合成腺苷酸琥珀酸合成酶基因dna链和/或其cdna链的pcr引物、或抗腺苷酸琥珀酸合成酶蛋白的抗体。
本发明的有益效果是:本发明提供了一种新的急性髓系白血病遗传和表观遗传标志物,该标志物相关性高。
附图说明
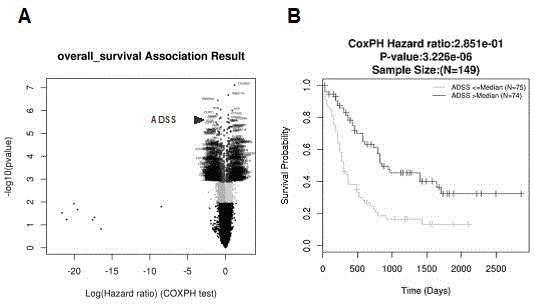
图1是图示tcga数据库分析发现与aml预后相关分子adss。
图2是图示aml患者低表达adss。
图3是图示adss低表达提示aml患者预后不良。a-d图中上面曲线表示adsslow,下面曲线表示adsshigh。
图4是ppi分析。
图5是adss高低表达组差异基因分析及富集分析。
具体实施方式
实施例1急性髓系白血病诊断和预后标志分子adss的筛选与验证
一、材料和方法
1生物信息学数据分析工具
1.1癌症和肿瘤基因图谱(thecancergenomeatlas,tcga)
tcga计划是由美国政府投资,旨在利用基因组测序技术,将人类的全部癌症基因组变异图谱绘制而出,并进行对比分析,研究rna和蛋白表达、dna拷贝数变化、与患者预后之间的关系。目前为止,tcga数据库中已收录了超过30种人类肿瘤数据,对于包含aml在内的肿瘤诊断与治疗研究具有重要意义。
基因表达汇编(geneexpressionomnibus,geo)数据库
geo是一个收录高通量基因表达和其他功能性基因组学数据的国际公用数据库,由美国国家医学图书馆国家生物技术信息中心支持和维护,于2000年创建,收集来自全球范围内的基因研究内容,其中接受并包含原始或者已加工的实验信息,样本数据信息,基因表达数据信息等。geo数据库不仅可以提供研究相关的信息,同时还拥有网络为基础的工具包、策略包,以帮助研究者收集、处理和分析数据,提高了信息的可视化。
string数据库
string数据库可应用于2000多个物种,包含上千万种蛋白质之间的相互作用关系和其功能联系等。
r语言(languager)
r语言是一种由rfoundation支持,用于统计学分析、编辑、绘图等的编程语言和自由开放的软件环境,在全基因组表达芯片及高通量测序的分析中具有广泛应用。
统计产品与服务解决方案(statisticalproductandservicesolutions,spss)软件
spss最大的优点是数据录入、整理、分析功能于一身,自由地按照统计学规范的方法处理缺失值、比较值,进行回归分析、聚类分析、生存分析、均值比较和统计性描述等,同时可以根据整理的数据绘制各种图形。
graphpadprism医学绘图数据分析工具
graphpadprism是一款基础生物统计及绘图综合软件,集曲线适配和科学图表绘制与一体,帮助科研工作者处理和分析实验结果等。
生物信息学数据分析方法
2.1tcga和geo数据库生存分析
利用tcga和geo数据库中aml患者预后数据信息,整合患者基因表达数据,使用survival包作kaplan-meier生存分析,并进行log-rank检验。得到一组与tcga数据库中aml患者生存相关的基因。按p值从小到大排列,选取与预后最具统计学相关性的前30位分子作为候选分子。
数据库筛选aml患者与正常人差异表达基因(differentiallyexpressedgenes,degs)分析
在geo数据库里寻找具有aml患者和正常对照组信息的数据集,通过r语言整理基因表达数据,比较aml患者与对照组全基因组表达水平,得到一系列差异表达基因。检索2.1中得出的30个候选分子表达差异水平,选取表达差异最具统计学意义的生存相关分子进行研究。
差异表达基因的单因素和多因素cox风险模型(coxproportional-hazardsmodel)分析
由于tcga数据库中包含较为全面的患者临床数据信息,所以选择使用tcga数据库里的信息进行单因素和多因素的cox回归分析。首先将数据库中的各临床特征如性别、年龄等作为单一因素,分别检验其与生存的相关性。选取对生存有影响的临床特征为变量,结合候选分子的表达进行多因素cox回归分析,探究候选分子表达水平是否可做为aml预后的独立影响因素。
目的基因表达与临床特征联系分析
根据目的基因表达量高低进行分组,统计各组的临床特征,进行卡方检验,探究可能与候选分子表达水平具有相关性的临床因素。
ppi分析
在string数据库中(https://string-db.org)对该目的基因构建相关蛋白互作网络,并对结果进行富集分析,包括geneontology(go)分析,kyotoencyclopediaofgenesandgenomes(kegg)pathway分析和diseaseontology(do)分析,观察这些蛋白可以被注释于哪些生物学功能通路和疾病谱中。将aml患者依据候选分子表达量由大到小的顺序进行排序,选取前25%作为高表达组,后25%作为低表达组,用r语言分析高低表达组患者的全基因组表达水平,得到差异基因,对差异性最具统计学意义的分子进行如上富集分析。
二、结果
1adss与aml患者的预后密切相关,并在aml骨髓样本中异常表达
表1tcga中与aml生存相关前30分子
表2tcga生存相关分子在geo数据库中表达差异分析
通过对tcga数据库中aml患者基因表达谱与生存信息的综合分析,得到一系列与aml患者预后相关的分子(见图1),从中选取与aml患者预后最密切相关的30个分子作为研究候选(见表1)。在geo数据库gse13159数据集中对上述分子进行aml患者与对照组的表达差异统计学分析,得出上述分子的基因表达差异倍数和p值,并按p值从小到大顺序排列(见表2),选取表达差异最显著的生存相关分子adss作为研究对象。
患者组低表达adss
在gse13159数据集中分析aml患者及正常对照者体内adss表达水平,发现aml组(n=501)相较对照组(n=73)低表达(见图2a)。根据2018年最新指南,将aml患者依据细胞遗传学特征进行危险度分层,比较不同风险度aml患者adss表达水平,结果显示细胞遗传学危险度越高的患者体内adss表达量越低(见图2b)。
低表达提示aml患者预后不良
将geo数据库(gse71014、gse106291)及tcga数据库中aml患者依据adss表达量高低分为两组,结合患者生存信息进行预后分析。结果显示,在各个数据集中,adss低表达组患者与高表达组相比具有更差的总生存(os)和无事件生存(efs)(见图3)。
可作为影响aml患者预后的独立因素
表3tcga数据库中adss多因素cox回归分析
对tcga数据库中各临床因素与生存信息进行单因素分析,确定与aml预后有相关性的6个因素,分别为年龄、白细胞数、细胞遗传学分型、dnmt3a突变、runx1突变以及tp53突变。将这6个因素与adss表达水平进行多因素cox回归分析,adss对应的p值<0.05,说明adss可作为影响aml患者预后的独立因素(见表3)。
与aml患者fab分型和细胞遗传学分层相关
表4aml患者adss表达与临床特征相关性检验
将tcga数据库中aml患者依据adss表达量的中位数分为高低两组,整合aml患者各项临床特征,进行卡方检验,发现adss表达水平与aml患者fab分型和细胞遗传学预后分层相关(见表4)。
可能通过影响腺苷酸合成参与aml发生
在string网站中对adss进行分析,发现adss可能与核苷酸代谢相关蛋白存在相互作用,从而影响疾病的发生发展(见图4)。将adss高表达组和低表达组患者的全基因组表达水平用r语言进行分析,得到差异表达基因,并对高低表达组差异基因进行富集分析,结果同样提示,adss可能通过影响核苷酸代谢参与aml发生与发展(见图5)。
综上所述,基于生物信息学分析adss在aml患者中的表达受抑,提示adss可能作为aml发生过程中的潜在抑癌基因。adss的低表达与多种aml患者的临床和生物学特征相关,可能是aml患者新的诊断及预后评估潜在分子。因此,本专利提示adss可能作为aml诊断、治疗、疾病进展监测及预后评估的靶点分子。
以上所述仅是本专利的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本专利技术原理的前提下,还可以做出若干改进和替换,这些改进和替换也应视为本专利的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!