基于双随机游走重启动的多数据整合环状RNA与疾病相关性预测方法与流程
本发明属于生物信息技术领域,具体涉及一种基于双随机游走重启动的多数据整合环状rna与疾病相关性预测方法。
背景技术:
最近,一种新的生物分子环状rna引起了人们的广泛关注。环状rna是一种相对新颖的生物分子,参与生物生命的各种活动并控制基因的表达。与拥有游离的3'端和5'端的线性rna不同,环状rna的结构是一个闭环结构,既没有游离在外的5’-cap端,也没有3’-polaydenylated尾端结构。第一个环状rna是在植物病毒中发现的。由于稳定的环结构和低表达水平,环状rna经常被鉴定为分子片段或转录的副产物。然而,随着高通量序列技术的发展,逐渐发现了越来越多的环状rna。同时,相关的生物学功能表明,在哺乳动物细胞中的环状rna具有内源性、丰富性、保守性和稳定性。许多证据表明,环状rna可分为四种类型:外显子环状rna由背部拼接的外显子组成;内含子环状rna主要来源于组(i,ii)内含子,内部套索和外源性trna内含子。外显子环状rna同时被外显子和内含子环化,而基因间环状rnas由两个内含子环状rna片段组成。越来越多的证据表明,环状rna在许多生物进程中发挥着重要的作用或功能。环状rnas也可以被认为是竞争性内源性rna或mirna的海绵,根据已有的研究证明,circ-sry,circ-hipk3,mm9_circ_012559都可被视为mirna海绵。同时,一些研究还表明环状rna可以与rna结合蛋白(rbps)相互作用。环状rna不仅可以调节基因转录过程,而且其中一些可以被翻译为蛋白质。
环状rnas除了对多种生物过程有影响外,还与不同复杂的疾病有关。环状rna具有一些独特的特征,如保守性、丰富性和组织特异性,这使得环状rna有可能成为疾病的标志物,特别是对一些肿瘤。根据环状rna在不同组织中的不同表达水平,我们可以确定正常人与患者之间的差异表达。因此,这些差异可以帮助我们预测或诊断疾病。由于qpcr技术,可以将肺癌中的环状rna表达特征与邻近的正常组织进行比较。环状rnacirs-7可以下调与肺相关的组织或细胞,而环状rna环状rna_100876和hsa_circ_0013958可以上调在肺对应的组织、细胞或血浆。然而,通过微阵列芯片技术,环状rna在胃相关组织和邻近胃的正常部位之间表现出显着差异,这表明环状rna可被视为胃癌诊断和进展的生物标志。环状rnacircpvt1和hsa_circ_0000096均可通过下调机制影响胃组织或细胞。更重要的是,环状rna可以作为mirna海绵或基因调节因子发挥作用,这也使得环状rna在直肠组织中具有不同的敏感性和特异性,可作为直肠癌诊断或治疗的生物标记物。环状rnahsa_circ_001569是mirnamir-145的海绵,可以促进其与组织的结合靶点在结肠直肠细胞中的表达。
为了进一步研究环状rna与其他生物分子之间的复杂关系并促进疾病诊断,建立了一些与环状rna相关的数据库。circbase数据库是最早的环状rna相关数据库之一,它提供了mrna在染色体上的定位、rna碱基序列、靶基因等。circrnadb数据库也是一种广泛使用的环状rna数据库,其中收集了大量的环状rna注释数据,这些数据是从基因组信息,外显子剪接,基因组序列中提取的。为了分析不同组织中的环状rna表达,建立了通过人血液外泌体提供的环状rna,lncrna和mrna信息的exorbase数据库。此外,采用rna-seq样本中的环状rna表达的circnet数据库系统地鉴定环状rna调控途径和组织特异性表达谱。此外,还有一些数据库提供了环状rna和疾病之间的关系信息。circ2traits利用环状rna-mirna关系,mirna-疾病关系和疾病-单核苷酸多态性(snps)关系来获取环状rna-疾病关系。最近,人们总是关注研究环状rna个体和单一疾病的关系。为了使疾病与环状rna关系的研究更有效,一些数据库通过从数千种文献中提取手动收集这些分散的环状rna-疾病关系的信息,例如circr2disease数据库,circrnadisease数据库和circ2disease数据库。
尽管高通量序列技术已经应用于环状rna与疾病关系的识别,但是存在一些不可忽略的限制。虽然这些技术能够以较高的准确率来提取环状rna-疾病关系,但是仍然是耗时且成本高的。更重要的是,用于预测潜在的环状rna-疾病潜在关系的计算方法较少是另一个主要动机。
技术实现要素:
为了克服上述现有技术的缺点,本发明的目的在于提供基于双随机游走重启动的多数据整合环状rna与疾病相关性预测方法,本发明通过整合多种环状rna相似性网络以及疾病相似性网络,分别在多数据融合的环状rna相似性网络和疾病相似性网络采用了随机游走重启动的方法,考虑环状rna相似性和疾病相似性同时对环状ran-疾病对的影响,避免了冷启动的问题,提高了环状rna-疾病相互作用关系预测准确率。
为了达到上述目的,本发明采用以下技术方案予以实现:
基于双随机游走重启动的多数据整合环状rna与疾病相关性预测方法,包括以下步骤:
(1)人类环状rna-疾病关系提取
将环状rna-疾病关系网络转换成一个无向图,环状rna-疾病之间的关系的邻接矩阵a,a(i,j)表示环状rna-疾病关系邻接矩阵a中的一对环状rna-疾病关系实体,如果环状rnac(i)和与疾病d(j)存在关系,则a(i,j)=1,否则a(i,j)=0;
(2)构建环状rna功能注释语义相似性网络
由环状rna的靶点基因相关基因本体数据构建环状rna功能注释语义相似性网络,根据从人类蛋白质参考数据库下载基因对应的本体数据,再将环状rna的靶点基因和从人类蛋白质参考数据库中处理好的基因及其对应的本体数据进行匹配,通过一种基于信息增益的方法来计算两个环状rna之间的功能注释相似性,从而构建环状rna功能注释语义相似度网络;
(3)构建环状rna结构相似性网络
通过python的一个工具包biopython中的needleman-wunsch序列比对算法计算每对环状rna之间的碱基序列相似性得分,为了统一相似性分数的数量级,对环状rna的结构相似性进行归一化,最终获得环状rna结构相似性网络;
(4)构建环状rna功能相似性网络
首先通过计算环状rna相关的一种疾病gt与一组疾病gt之间的最大相似性得分,其被定义为smax(gt,gt),然后通过计算得出的某一疾病与全部疾病集合的最大相似性得分,计算两个环状rna之间的功能相似性,从而构建环状rna功能相似性网络;
(5)构建疾病语义相似性网络
将筛选出来的疾病在diseaseontology数据库中进行手动匹配,将疾病名称对应为相关的doid,其次采用名为dose的r包来计算每两种疾病之间的语义相似度得分,在得到每对疾病相似性得分之后进而可以构建疾病语义相似网络dsn1,疾病语义相似网络dsn1中的dsn1(i,j)表示疾病i和j的语义相似性得分;
(6)构建疾病功能相似性网络
在disgenet和人类在线孟德尔遗传数据库中下载疾病相关的基因数据,通过统计学算法jaccard来计算疾病功能相似性,进而构建疾病功能相似网络dsn2,疾病功能相似网络dsn2中的dsn2(i,j)代表疾病i和j的功能相似性分数;
(7)整合环状rna相似性网络
通过步骤(2)、(3)以及(4)构建的环状rna功能注释语义相似网络csn1,结构相似性网络csn2以及功能相似性网络csn3,整合环状rna相似性网络:
(8)整合疾病相似性网络
将构建的疾病语义相似性网络dsn1和疾病功能相似性网络dsn2整合成最终的疾病相似性网络dsn;
(9)通过双随机游走算法预测环状rna-疾病潜在关系
为了给整合后的环状rna相似性网络和疾病相似性网络中的环状rna和疾病节点赋予一个初始的传播概率,将环状rna相似性网络和疾病相似性网络按照列来进行标准化,ncs(i,j)和nds(i,j)分别代表的是标准化后的环状rnai和j之间的相似性得分以及疾病i和j的相似性得分,为了在整合后的环状rna相似性网络和疾病相似网络中进行节点概率传播的概率转移,首先初始化整合后环状rna相似性网络和疾病相似性网络中的环状rna和疾病节点的转移概率,再通过分别在整合后的环状rna相似网络和疾病相似性网络中采用随机游走算法,最后综合在环状rna相似性网络和疾病相似性网络中的预测结果获取最终的环状rna-疾病关系预测结果。
进一步地,步骤(2)中按式(1)计算环状rnac(i)和c(j)之间的相似性得分:
式中csn1(i,j)表示环状rnac(i)和c(j)之间的相似性分数,pro(ci)和pro(cj)分别表示环状rnac(i)/c(j)靶点基因相关的本体数据的数量与所有与靶点基因相关的本体数据总数量之间的比例,pro(ci∪cj)代表环状rnaci和cj靶点基因共同相关的本体数据数量和所有靶点基因相关的本体数据总数的比例。
进一步地,步骤(3)中按式(2)对环状rna的结构相似性进行归一化:
式中csn2代表的是经过归一化处理的环状rna结构相似性网络,csn2(i,j)代表的是环状rnac(i)和c(j)的相似性分数,式中nws(c(i),c(j))代表环状rnac(i)和c(j)之间的needleman-wunsch序列比对算法得出的相似性得分。
进一步地,步骤(4)中按式(3)计算一种疾病gt与一组疾病gt之间的最大相似性得分smax(gt,gt):
通过式(3)计算得出的某一疾病与疾病集合的最大相似性得分,按式(4)来计算两个环状rna之间的功能相似性:
式中csn3代表环状rna功能相似性网络,其中csn3(i,j)表示环状rnaci和cj的功能相似性得分,gti和gtj分别代表的是环状rnaci和cj相关疾病集合,gtil和gtjq分别表示gti和gtj疾病集合中的某一疾病,n和m分别代表环状rnaci和cj相关疾病的数量。
进一步地,步骤(6)中按式(5)计算疾病i和j的功能相似性分数:
式中dg(i)和dg(j)分别表示疾病i和j相关基因集合。
进一步地,步骤(7)中按式(6)来整合环状rna相似性网络:
式中csn表示整合后的环状rna相似性网络,其中csn(i,j)代表环状rnai和j的相似性得分。
进一步地,步骤(8)中按式(7)来计算dsn(i,j):
dsn(i,j)=αdsn1(i,j)+(1-α)dsn2(i,j)式(7)
式中α代表疾病相似性整合调和平均参数,dsn(i,j)代表整合后的疾病i和j的相似性得分。
进一步地,步骤(9)中ncs(i,j)和nds(i,j)按式(8)和式(9)计算:
为了在整合后的环状rna相似性网络和疾病相似网络中进行节点概率传播的概率转移,分别按式(10)和式(11)计算环状rna和疾病的转移概率:
crt=β*ncs*crt-1+(1-β)a式(10)
drt=β*nds*drt-1+(1-β)a式(11)
式中crt和drt分别表示每一次迭代后的环状rna和疾病网络上的随机游走的每对环状rna-疾病潜在关系得分,β表示在每次随机游走迭代过程中的衰减因子,t是环状rna和疾病网络上随机游走的迭代次数,在迭代完成后能获得每一对环状rna-疾病关系的可能性得分。
进一步地,步骤(9)中的具体迭代方法如下:
step1:初始化环状rna相似性网络和疾病相似性网络迭代标志lflag=0以及rflag=0;
step2:判断当前迭代次数小于或等于给定的在环状rna相似性网络中的迭代次数,则通过式(10)来进行概率传播,并将环状rna相似性网络的迭代标志lflag设为1;
step3:判断当前迭代次数小于或等于给定的在疾病相似性网络中的迭代次数,则通过式(11)来进行概率传播,并将疾病相似性网络的迭代标志rflag设为1;
step4:根据式(12)计算当且迭代中的环状rna-疾病潜在关系得分:
rw=(lflag*cr+rflag*dr)/(lflag+rflag)式(12)
式中rw表示的是本轮迭代中每对环状rna-疾病相关性预测得分,cr代表的是随机游走算法在整合后的环状rna相似性网络中的环状rna-疾病相关性预测得分,dr代表的是随机游走算法在整合后的疾病相似性网络中的环状rna-疾病相关性预测得分;
step5:判断迭代次数是否等于给定的环状rna相似性网络迭代次数icsn和疾病相似性网络迭代次数idsn中的最大的值,若是,迭代完成;否则,回到step1。
与现有技术相比,本发明具有以下有益效果:
1、本发明通过环状rna靶点基因相关本体数据,碱基序列数据以及相关疾病的语义相似性建立环状rna功能注释语义相似性,结构相似性以及功能相似性网络,通过疾病相关基因和表型数据建立疾病功能相似性和语义相似性网络。再将多网络整合成一个最终的环状rna相似性网络和疾病相似性网络,考虑多方面数据,与其他关系预测方法相比,多元相似性网络的综合考虑,减少了信息的损失,尽可能的在整合的多数据网络中挖出潜在的环状rna-疾病潜在关系,提高了环状rna-疾病关系的预测准确率。
2、本发明在通过分别在环状rna相似网络中和疾病相似网络中采用随机游走重启动算法的方法,充分的考虑了环状rna相似性和疾病相似性对潜在关系的影响,减少了冷启动问题,提高了潜在环状rna-疾病关系预测的准确率。
3、采用本发明能够有效地预测出存在潜在关系的环状rna-疾病关系,为进一步分析环状rna的生物学意义以及环状rna与疾病之间的复杂关系奠定了基础。此研究不仅有助于理解细胞的运作机理和生命活动机制,也为探讨重大疾病的机理、疾病的诊断、临床治疗、预防以及新药物的开发提供前期的理论分析,这将为合成生物学与系统医学的研究与发展提供重要的理论指导和应用价值。
附图说明
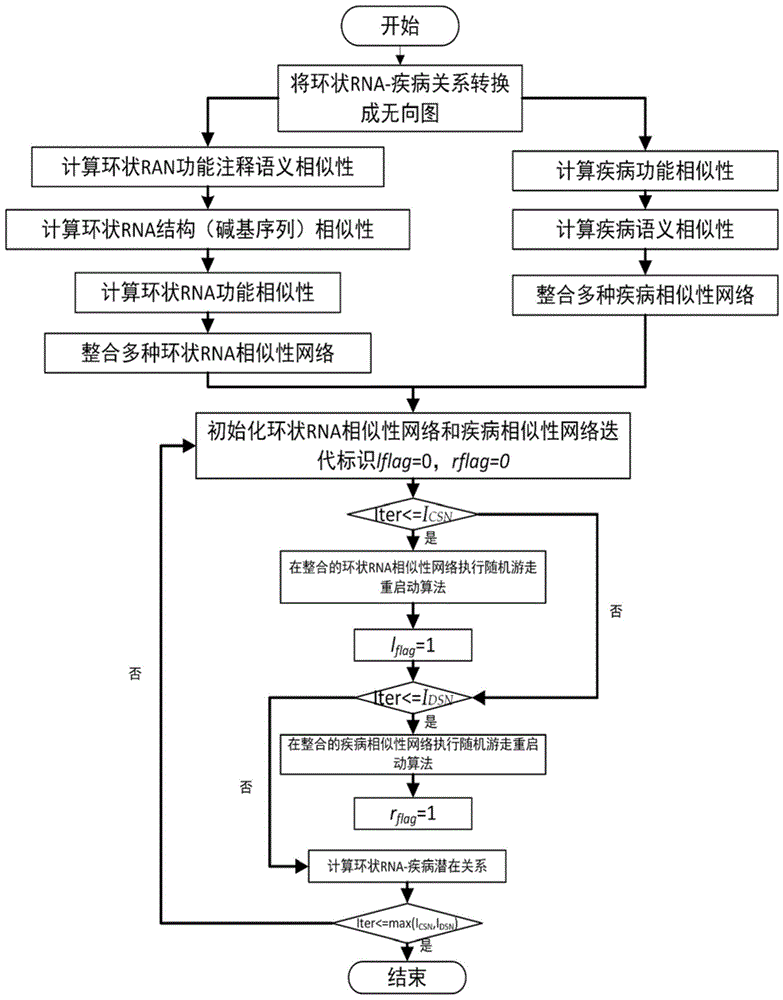
图1为本发明的基于双随机游走重启动的多数据整合环状rna与疾病相关性预测方法流程示意图。
具体实施方式
为了使本技术领域的人员更好地理解本发明方案,下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分的实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都应当属于本发明保护的范围。
需要说明的是,本发明的说明书和权利要求书及上述附图中的术语“第一”、“第二”等是用于区别类似的对象,而不必用于描述特定的顺序或先后次序。应该理解这样使用的数据在适当情况下可以互换,以便这里描述的本发明的实施例能够以除了在这里图示或描述的那些以外的顺序实施。此外,术语“包括”和“具有”以及他们的任何变形,意图在于覆盖不排他的包含,例如,包含了一系列步骤或单元的过程、方法、系统、产品或设备不必限于清楚地列出的那些步骤或单元,而是可包括没有清楚地列出的或对于这些过程、方法、产品或设备固有的其它步骤或单元。
下面结合附图对本发明做进一步详细描述:
如图1所示,本发明基于双随机游走重启动的多数据整合环状rna与疾病相关性预测方法,为了达到较好的潜在环状rna-疾病相互作用关系预测效果,环状rna功能注释语义相似,结构相似以及功能相似性网络被用来构建多元数据整合的环状rna相似性网络;构建疾病功能相似和语义相似网络来整合疾病相似性网络。再通过概率传播算法(基于整合的环状rna相似网络和疾病的相似性网络的双随机游走重启动方法),解决了冷启动问题。从而提高了潜在环状rna-疾病关系预测的准确性。
具体包括以下步骤:
1)人类环状rna-疾病关系提取
将环状rna-疾病关系网络转换成一个无向图,环状rna-疾病之间的关系的邻接矩阵a,a(i,j)可表示为一对环状rna-疾病关系实体,如果环状rnac(i)和与疾病d(j)存在关系,a(i,j)=1,否则a(i,j)=0;
2)构建环状rna功能注释语义相似性网络
由环状rna的靶点基因相关基因本体数据构建环状rna功能注释语义相似性网络。根据从人类蛋白质参考数据库(hprd)下载基因对应的本体数据,再将环状rna的靶点基因和从hprd中处理好的基因及其对应的本体数据进行匹配,通过一种基于信息增益的方法来计算两个环状rna之间的功能注释相似性,从而构建环状rna功能注释语义相似度网络(csn1),按式(1)计算环状rnac(i)和c(j)之间的相似性得分:
式中csn1(i,j)表示环状rnac(i)和c(j)之间的相似性分数,pro(ci)和pro(cj)分别表示环状rnac(i)/c(j)靶点基因相关的本体数据的数量与所有与靶点基因相关的本体数据总数量之间的比例。pro(ci∪cj)代表环状rnaci和cj靶点基因共同相关的本体数据数量和所有靶点基因相关的本体数据总数的比例;
3)构建环状rna结构相似性网络
通过python的一个工具包biopython中的needleman-wunsch序列比对算法来计算每对环状rna之间的碱基序列相似性得分,为了统一相似性分数的数量级,按式(2)对环状rna的结构相似性进行归一化:
式中csn2代表的是经过归一化处理的环状rna结构相似性网络,csn2(i,j)代表的是环状rnai和j的相似性分数,式中nws(c(i),c(j))代表环状rnac(i)和c(j)之间的needleman-wunsch序列比对算法得出的相似性得分;
4)构建环状rna功能相似性网络
首先通过计算一种疾病gt与一组疾病gt之间的最大相似性得分,其被定义为smax(gt,gt),按式(3)计算:
通过式(3)计算得出的某一疾病与疾病集合的最大相似性得分,按式(4)来计算两个环状rna之间的功能相似性:
式中csn3代表环状rna功能相似性网络,其中csn3(i,j)表示环状rnaci和cj的功能相似性得分。gti和gtj分别代表的是环状rnaci和cj相关疾病集合。gtil和gtjq分别表示gti和gtj疾病集合中的某一疾病。n和m分别代表环状rnaci和cj相关疾病的数量;
5)构建疾病语义相似性网络
将筛选出来的疾病在diseaseontology数据库中进行手动匹配,将疾病名称对应为相关的doid。其次采用名为dose的r包来计算每两种疾病之间的语义相似度得分。dsn1代表疾病语义相似性网络,其中的dsn1(i,j)表示疾病i和j的语义相似性得分;
6)构建疾病功能相似性网络
鉴于还应考虑疾病功能特征的信息,还需要在disgenet和人类在线孟德尔遗传数据库(omim)中下载疾病相关的基因数据,通过统计学算法jaccard来计算疾病功能相似性,进而构建疾病功能相似网络dsn2,dsn2(i,j)代表疾病i和j的功能相似性分数,按式(5)计算:
式中dg(i)和dg(j)分别表示疾病i和j相关基因集合;
7)整合环状rna相似性网络
通过步骤(2)、(3)以及(4)构建的环状rna功能注释语义相似网络csn1,结构相似性网络csn2以及功能相似性网络csn3,按(6)来整合环状rna相似性网络:
式中csn表示整合后的环状rna相似性网络,其中csn(i,j)代表环状rnai和j的相似性得分;
8)整合疾病相似性网络
将构建的疾病语义dsn1和功能相似性dsn2网络整合成最终的疾病相似性网络dsn,按式(7)来计算dsn(i,j):
dsn(i,j)=αdsn1(i,j)+(1-α)dsn2(i,j)式(7)
式中α代表疾病相似性整合调和平均参数,dsn(i,j)代表整合后的疾病i和j的相似性得分;
9)通过双随机游走算法预测环状rna-疾病潜在关系
为了给整合后的环状rna相似性网络和疾病相似性网络中的环状rna和疾病节点赋予一个初始的传播概率,将环状rna和疾病相似性网络的按照列来进行标准化,ncs(i,j)和nds(i,j)分别代表的是归一化后的环状rnai和j之间的相似性得分以及疾病i和j的相似性得分,ncs(i,j)和nds(i,j)按式(8)和式(9)计算:
为了在整合后的环状rna相似性网络和疾病相似网络中进行节点概率传播的概率转移,分别按式(10)和式(11)计算环状rna和疾病的转移概率:
crt=β*ncs*crt-1+(1-β)a式(10)
drt=β*nds*drt-1+(1-β)a式(11)
式中crt和drt分别表示每一次迭代后的环状rna和疾病网络上的随机游走的每对环状rna-疾病潜在关系得分,β表示在每次随机游走迭代过程中的衰减因子,t是环状rna和疾病网络上随机游走的迭代次数。在迭代完成后能获得每一对环状rna-疾病关系的可能性得分。
本发明步骤(9)中的具体迭代方法如下:
step1:初始化环状rna相似性网络和疾病相似性网络迭代标志lflag=0以及rflag=0;
step2:判断当前迭代次数小于或等于给定的在环状rna相似性网络中的迭代次数,就通过权利要求1所述的,其特征在于,步骤(9)中的式(10)来进行概率传播,并将环状rna相似性网络的迭代标志lflag设为1;
step3:判断当前迭代次数小于或等于给定的在疾病相似性网络中的迭代次数,就通过权利要求1所述的,其特征在于,步骤(9)中的式(11)来进行概率传播,并将疾病相似性网络的迭代标志rflag设为1;
step4:根据式(12)计算当前迭代中的环状rna-疾病潜在关系得分:
rw=(lflag*cr+rflag*dr)/(lflag+rflag)式(12)
step5:判断迭代次数是否等于给定的最大的环状rna相似性网络/疾病相似性网络迭代次数icsn和idsn,迭代完成;否则,回到step1。
以下通过具体实施例对本发明进一步详细说明:
下面是以circr2disease数据库中的环状rna-疾病关系为例的一种基于双随机游走重启动的多数据整合环状rna与疾病相关性预测方法,具体操作如下:
本实施例以采自circr2disease数据库提供的环状rna-疾病关系数据作为仿真数据集,根据环状rna靶点基因相关的本体数据(从人类蛋白质参考数据库(hprd)中获取),碱基序列数据(circbase数据库中下载)以及相关疾病语义相似性数据和疾病相关基因数据(从disgenet数据库和人类在线孟德尔遗传数据库(omim)数据库中获取)以及表型数据(在diseaseontology数据中,将疾病转换成对应的doid),从circr2diseas数据库中的提供的739对已知环状rna-疾病相似性数据,筛选出200个环状rna,42个疾病数据以及212对环状rna-疾病关系数据。实验平台为windows10操作系统,intel酷睿i5-7400双核3.00ghz处理器,8gb物理内存,用pycharm2017软件实现本发明的方法。
具体步骤如下:
1、人类环状rna-疾病关系提取
将包含了200个环状rna和42个疾病的拥有212个环状rna-疾病关系网络转换成一个无向图,环状rna-疾病之间的关系的邻接矩阵a,a(i,j)可表示为一对环状rna-疾病关系实体,如果环状rnac(i)和与疾病d(j)存在关系,a(i,j)=1,否则a(i,j)=0;
2、构建环状rna功能注释语义相似性网络
由200个环状rna的靶点基因相关基因本体数据构建环状rna功能注释语义相似性网络。根据从人类蛋白质参考数据库(hprd)下载基因对应的本体数据,再将环状rna的靶点基因和从hprd中处理好的基因及其对应的本体数据进行匹配,通过一种基于信息增益的方法来计算两个环状rna之间的功能注释相似性,从而构建环状rna功能注释语义相似度网络(csn1),由式(1)计算环状rnac(i)和c(j)之间的相似性得分:
式中csn1(i,j)表示环状rnac(i)和c(j)之间的相似性分数,pro(ci)和pro(cj)分别表示环状rnac(i)/c(j)靶点基因相关的本体数据的数量与所有与靶点基因相关的本体数据总数量之间的比例。pro(ci∪cj)代表环状rnaci和cj靶点基因共同相关的本体数据数量和所有靶点基因相关的本体数据总数的比例;
3、构建环状rna结构相似性网络
通过python的一个工具包biopython中的needleman-wunsch序列比对算法来计算200个环状rna之间的碱基序列相似性得分,为了统一相似性分数的数量级,按式(2)对环状rna的结构相似性进行归一化:
式中csn2代表的是经过归一化处理的环状rna结构相似性网络,csn2(i,j)代表的是环状rnai和j的相似性分数,式中nws(c(i),c(j))代表环状rnac(i)和c(j)之间的needleman-wunsch序列比对算法得出的相似性得分;
4、构建环状rna功能相似性网络
首先通过计算一种疾病gt与一组疾病gt之间的最大相似性得分,其被定义为smax(gt,gt),按式(3)计算:
通过式(3)计算得出的某一疾病与疾病集合的最大相似性得分,按式(4)来计算两个环状rna之间的功能相似性:
式中csn3代表环状rna功能相似性网络,其中csn3(i,j)表示环状rnaci和cj的功能相似性得分。gti和gtj分别代表的是环状rnaci和cj相关疾病集合。gtil和gtjq分别表示gti和gtj疾病集合中的某一疾病。n和m分别代表环状rnaci和cj相关疾病的数量;
5、构建疾病语义相似性网络
将筛选出来的42个疾病在diseaseontology数据库中进行手动匹配,将疾病名称对应为相关的doid。其次采用名为dose的r包来计算每两种疾病之间的语义相似度得分。dsn1代表疾病语义相似性网络,其中的dsn1(i,j)表示疾病i和j的语义相似性得分;
6、构建疾病功能相似性网络
鉴于还应考虑疾病功能特征的信息,还需要在disgenet和人类在线孟德尔遗传数据库(omim)中下载42个疾病相关的基因数据,通过统计学算法jaccard来计算疾病功能相似性,进而构建疾病功能相似网络dsn2,dsn2(i,j)代表疾病i和j的功能相似性分数,按式(5)计算:
式中dg(i)和dg(j)分别表示疾病i和j相关基因集合;
7、整合环状rna相似性网络
通过步骤(2)、(3)以及(4)构建的环状rna功能注释语义相似网络csn1,结构相似性网络csn2以及功能相似性网络csn3,按(6)来整合环状rna相似性网络:
式中csn表示整合后的环状rna相似性网络,其中csn(i,j)代表环状rnai和j的相似性得分;
8、整合疾病相似性网络
将构建的疾病语义dsn1和功能相似性dsn2网络整合成最终的疾病相似性网络dsn,按式(7)来计算dsn(i,j):
dsn(i,j)=αdsn1(i,j)+(1-α)dsn2(i,j)式(7)
式中α代表疾病相似性整合调和平均参数,dsn(i,j)代表整合后的疾病i和j的相似性得分;
9、通过双随机游走算法预测环状rna-疾病潜在关系
为了给整合后的环状rna相似性网络和疾病相似性网络中的环状rna和疾病节点赋予一个初始的传播概率,将环状rna和疾病相似性网络的按照列来进行标准化,ncs(i,j)和nds(i,j)分别代表的是归一化后的环状rnai和j之间的相似性得分以及疾病i和j的相似性得分,ncs(i,j)和nds(i,j)按式(8)和式(9)计算:
为了在整合后的环状rna相似性网络和疾病相似网络中进行节点概率传播的概率转移,分别按式(10)和式(11)计算环状rna和疾病的转移概率:
crt=β*ncs*crt-1+(1-β)a式(10)
drt=β*nds*drt-1+(1-β)a式(11)
式中crt和drt分别表示每一次迭代后的环状rna和疾病网络上的随机游走的每对环状rna-疾病潜在关系得分,β表示在每次随机游走迭代过程中的衰减因子,t是环状rna和疾病网络上随机游走的迭代次数。在迭代完成后能获得每一对环状rna-疾病关系的可能性得分。
本发明步骤(9)中的具体迭代方法如下:
step1:初始化环状rna相似性网络和疾病相似性网络迭代标志lflag=0以及rflag=0;
step2:判断当前迭代次数小于或等于给定的在环状rna相似性网络中的迭代次数,就通过权利要求1所述的,其特征在于,步骤(9)中的式(10)来进行概率传播,并将环状rna相似性网络的迭代标志lflag设为1;
step3:判断当前迭代次数小于或等于给定的在疾病相似性网络中的迭代次数,就通过权利要求1所述的,其特征在于,步骤(9)中的式(11)来进行概率传播,并将疾病相似性网络的迭代标志rflag设为1;
step4:根据下式计算当且迭代中的环状rna-疾病潜在关系得分:
rw=(lflag*cr+rflag*dr)/(lflag+rflag)
step5:判断迭代次数等于给定的最大的环状rna相似性网络/疾病相似性网络迭代次数icsn和idsn分别设置为3和1,迭代完成;否则,回到step1。
为了验证本发明的有效性,发明人采用本发明实施例1基于双随机游走的多数据整合环状rna与疾病相关性预测方法对circr2disease数据库中的环状rna-疾病关系进行预测,对环状rna-疾病关系网络中的已知的作用关系实施留一交叉验证,以此来分析本发明的预测性能,结果见表1-3,表1显示了与当前其他关系预测方法预测的环状rna-疾病潜在关系的结果进行预测精确率(precision)、召回率(recall)、准确率(accuracy)以及f1-measure的比较,表2显示了通过5折交叉验证将本发明方法与其它关系预测方法在10个典型疾病与环状rna关系预测结果在获取的auc值的比较。表3列出了本发明预测出的前10个潜在环状rna-膀胱癌关系在其他数据库或相关文献中的验证。
表1显示了采用本发明预测出的环状rna-疾病关系与circr2disease数据中标准数据进行比较的精确率(precision)、召回率(recall)、准确率(accuracy)以及f1-measure的比较,以及与其它10种关系预测方法的预测结果的比较。由表1可以看出,与其他方法相比,本发明方法能更有效地预测环状rna-疾病潜在关系,本发明方法都有最高的预测精确率(precision),召回率(recall)、准确率(accuracy)以及f1-measure。表2显示了通过对10个典型疾病的环状rna进行5折交叉验证,利用预测出的环状rna-疾病的结果,计算出对应的auc结果。由表2可以看出,与其他方法相比,本发明在对于这10中典型疾病与环状rna预测结果中表明,预测结果最好。表3本发明预测出的前10个潜在环状rna-膀胱癌关系在其他数据库或相关文献中的验证情况,由表3可以看出在预测的前十个和乳腺癌相关的环状rna中,被其他数据库和相关文献验证的环状rna有9个,’circrnabcrc4/hsa_circ_001598/hsa_circ_0001577’是一个被预测为的膀胱癌潜在相关的环状rna。由表1、表2和表3可以看出,本发明能够准确、有效地预测出潜在的环状rna-疾病关系。
表1本发明与其他关系预测方法预测环状rna-疾病关系在准确率上的比较
表2本发明与其他关系预测方法预测环状10个典型疾病-环状rna关系auc值的比较
表3本发明预测出的潜在环状rna-膀胱癌癌关系在其他数据库/文献中的验证
综上所述,本发明基于双随机游走的多数据整合环状rna与疾病相关性预测方法,通过将环状rna-疾病关系网络转化为无向图、计算环状rna功能注释语义相似性、结构相似性以及功能相似性,计算疾病功能以及语义相似性,将多种环状rna相似性网络和疾病相似性网络整合成综合的环状rna相似性网络以及疾病相似性网络,将随机游走重启动算法分别应用在整合后的环状rna相似性网以及疾病相似性网络,避免冷启动问题,预测潜在环状rna-疾病关系。本发明方法能准确地预测出潜在环状rna-疾病关系;仿真实验结果表明,精确度、召回率、准确度、f1-measure等指标较优;与其他关系预测方法相比,将多数据融合的相似性网络中采用双随机游走重启动算法预测环状rna-疾病潜在关系,提高了环状rna-疾病关系的预测准确率。
以上内容仅为说明本发明的技术思想,不能以此限定本发明的保护范围,凡是按照本发明提出的技术思想,在技术方案基础上所做的任何改动,均落入本发明权利要求书的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!