用于检测和抑制由融合事件引起的比对错误的方法与流程
用于检测和抑制由融合事件引起的比对错误的方法
[0001]
交叉引用
[0002]
本国际专利申请要求于2018年4月13日提交的美国临时专利申请第62/657,200号的优先权,该美国临时专利申请通过引用以其整体并入本文。
[0003]
背景
[0004]
由基因组重排事件引起的重复的基因组区域(duplicated genomic regions)可能对临床测序应用中的准确变异识别(calling)构成挑战,因为重复特异性变异可能被错误地分配给靶。加工的假基因(ppg)是重复的编码序列的一个来源,该重复的编码序列可来源于line(长散在元件(long interspersed elements))介导的加工的mrna(缺少内含子序列)的逆转录和基因组整合,导致原始基因的部分或完整拷贝。由参考基因组中发现的假基因导致的假阳性变异(诸如,pik3ca和pten的假基因)已经得到了很好的研究;然而,罕见且甚至是个体特异性的癌症相关的ppg的发现表明需要在逐个样品的基础上对与ppg相关的临床假象进行更系统的审查和调整。
[0005]
概述
[0006]
在某些方面,本公开内容提供了一种用于检测基因序列读段(genetic sequence reads)中的比对错误的方法,该方法包括:对来自受试者的样品的无细胞脱氧核糖核酸(dna)分子进行测序,其中每个无细胞dna分子产生多于一个序列读段;将源自测序的序列读段与参考序列比对,以产生比对的序列读段;从比对的序列读段中鉴定包含基因内融合断点的基因融合读段的集合;以及通过鉴定在包含基因内融合断点的区域内包含遗传变异(genetic variants)的基因融合读段的一个或更多个的子集来检测比对错误,其中该区域包含与基因内融合断点相邻的一个或更多个核苷酸。
[0007]
在某些方面,本公开内容提供了一种用于在检测来自受试者的样品的无细胞dna分子中的真正遗传变异时抑制比对错误的方法,该方法包括:对来自受试者的样品的无细胞dna分子进行测序,其中每个无细胞dna分子产生多于一个序列读段;将源自测序的序列读段与参考序列比对,以产生比对的序列读段;从比对的序列读段中鉴定包含基因内融合断点的基因融合读段的集合;通过鉴定在包含基因内融合断点的区域内包含遗传变异的基因融合读段的一个或更多个的子集来检测比对错误,其中该区域包含与基因内融合断点相邻的一个或更多个核苷酸;过滤掉一个或更多个基因融合读段的子集中的一个或更多个检测到的比对错误的至少一部分,以产生过滤的序列读段;以及检测与参考序列相比包含真正遗传变异的过滤的序列读段。
[0008]
在某些方面,本公开内容提供了一种用于在检测来自受试者的样品的无细胞dna分子中的真正遗传变异时抑制比对错误的方法,该方法包括:对来自受试者的样品的无细胞dna分子进行测序,其中每个无细胞dna分子产生多于一个序列读段;将源自测序的序列读段与参考序列比对,以产生比对的序列读段;从比对的序列读段中鉴定包含基因内融合断点的基因融合读段的集合;通过鉴定包含遗传变异的基因融合读段的一个或更多个的子集来检测比对错误,其中该基因融合读段的一个或更多个的子集包括对应于smad4和/或raf1的基因序列;过滤掉基因融合读段的一个或更多个的子集中的一个或更多个检测到的
比对错误的至少一部分,以产生过滤的序列读段;以及检测与所述参考序列相比包含真正遗传变异的过滤的序列读段。
[0009]
在某些方面,本公开内容提供了一种用于检测基因序列读段中的比对错误的方法,该方法包括:对来自受试者的样品的无细胞dna分子进行测序,其中每个无细胞dna分子产生多于一个序列读段;将源自测序的序列读段与参考序列比对,以产生比对的序列读段;从比对的序列读段中确定包含基因内融合断点的基因融合读段的集合;确定在包含基因内融合断点的区域内包含遗传变异的基因融合读段的一个或更多个的子集,其中该区域包含与基因内融合断点相邻的一个或更多个核苷酸;以及将该区域内满足预定标准的每个遗传变异鉴定为比对错误。
[0010]
在某些方面,本公开内容提供了一种用于在检测来自受试者的样品的无细胞dna分子中的真正遗传变异时抑制比对错误的方法,该方法包括:对来自受试者的样品的无细胞dna分子进行测序,其中每个无细胞dna分子产生多于一个序列读段;将源自测序的序列读段与参考序列比对以产生比对的序列读段;从比对的序列读段中确定包含基因内融合断点的基因融合读段的集合;确定在包含基因内融合断点的区域内包含遗传变异的基因融合读段的一个或更多个的子集,其中该区域包含与基因内融合断点相邻的一个或更多个核苷酸;将该区域内满足预定标准的每个遗传变异鉴定为比对错误;过滤掉一个或更多个基因融合读段的子集中的一个或更多个比对错误,以产生过滤的序列读段;以及检测与参考序列相比包含真正遗传变异的过滤的序列读段。
[0011]
在某些方面,本公开内容提供了一种用于至少部分地使用计算机检测基因序列读段中的比对错误的方法,该方法包括:通过计算机接收序列信息,所述序列信息包括从来自受试者的生物样品中的无细胞核酸分子获得的基因序列读段;将基因序列读段与参考序列比对以产生比对的序列读段;从比对的序列读段中鉴定包含基因内融合断点的基因融合读段的集合;以及通过鉴定在包含基因内融合断点的区域内包含遗传变异的基因融合读段的一个或更多个的子集来检测比对错误,其中该区域包含与基因内融合断点相邻的一个或更多个核苷酸。
[0012]
在某些方面,本公开内容提供了一种用于至少部分地使用计算机在检测来自受试者的生物样品的无细胞核酸分子中的真正遗传变异时抑制比对错误的方法,该方法包括:通过计算机接收序列信息,所述序列信息包括从无细胞核酸分子获得的序列读段;将序列读段与参考序列比对以产生比对的序列读段;从比对的序列读段中鉴定包含基因内融合断点的基因融合读段的集合;通过鉴定在包含基因内融合断点的区域内包含遗传变异的基因融合读段的一个或更多个的子集来检测比对错误,其中该区域包含与基因内融合断点相邻的一个或更多个核苷酸;过滤掉一个或更多个基因融合读段的子集中的一个或更多个检测到的比对错误的至少一部分,以产生过滤的序列读段;以及检测与参考序列相比包含真正遗传变异的过滤的序列读段。
[0013]
在某些方面,本公开内容提供了一种用于至少部分地使用计算机在检测来自受试者的样品的无细胞核酸分子中的真正遗传变异时抑制比对错误的方法,该方法包括:通过计算机接收序列信息,所述序列信息包括从无细胞核酸分子获得的测序读段;将序列读段与参考序列比对以产生比对的序列读段;从比对的序列读段中鉴定包含基因内融合断点的基因融合读段的集合;通过鉴定包含遗传变异的基因融合读段的一个或更多个的子集来检
测比对错误,其中该基因融合读段的一个或更多个的子集包括对应于smad4、tyro3和/或raf1的基因序列;过滤掉基因融合读段的一个或更多个的子集中的一个或更多个检测到的比对错误的至少一部分,以产生过滤的序列读段;以及检测与所述参考序列相比包含真正遗传变异的过滤的序列读段。
[0014]
在某些方面,本公开内容提供了一种用于至少部分地使用计算机检测基因序列读段中的比对错误的方法,该方法包括:通过计算机接收序列信息,所述序列信息包括从来自受试者的生物样品中的无细胞核酸分子获得的基因序列读段;将基因序列读段与参考序列比对以产生比对的序列读段;从比对的序列读段中确定包含基因内融合断点的基因融合读段的集合;确定在包含基因内融合断点的区域内包含遗传变异的基因融合读段的一个或更多个的子集,其中该区域包含与基因内融合断点相邻的一个或更多个核苷酸;以及将该区域内满足预定标准的每个遗传变异鉴定为比对错误。
[0015]
在某些方面,本公开内容提供了一种用于至少部分地使用计算机在检测来自受试者的样品的无细胞核酸分子中的真正遗传变异时抑制比对错误的方法,该方法包括:通过计算机接收序列信息,所述序列信息包括从无细胞核酸分子获得的测序读段;将序列读段与参考序列比对以产生比对的序列读段;从比对的序列读段中鉴定包含基因内融合断点的基因融合读段的集合;通过鉴定包含遗传变异的基因融合读段的一个或更多个的子集来检测比对错误,其中该基因融合读段的一个或更多个的子集包括对应于smad4、tyro3和/或raf1的基因序列;过滤掉基因融合读段的一个或更多个的子集中的一个或更多个检测到的比对错误的至少一部分,以产生过滤的序列读段;以及检测与所述参考序列相比包含真正遗传变异的过滤的序列读段。
[0016]
在某些实施方案中,基因融合读段的集合对应于一个或更多个加工的假基因(ppg)。在某些实施方案中,一个或更多个ppg包括一个或更多个样品特异性ppg。在某些实施方案中,由于参考基因组中的缺口或者因为一个或更多个ppg是样品特异性ppg,因此一个或更多个ppg不存在于参考基因组中。在某些实施方案中,一个或更多个样品特异性ppg在受试者的群体中鉴定受试者。在某些实施方案中,一种或更多种ppg源自由以下组成的组中的基因的外显子序列:smad4、gnas、tp53、raf1、cdk4、tyro3、mapk1、stk11、ccnd1、hras、met、myc和nras。在某些实施方案中,一个或更多个ppg包括两个或更多个ppg,所述两个或更多个ppg源自由以下组成的组的一个或更多个序列:smad4、gnas、tp53、raf1、cdk4、tyro3、mapk1、stk11、ccnd1、hras、met、myc和nras。在某些实施方案中,一个或更多个ppg包括三个或更多个ppg,所述三个或更多个ppg源自由以下组成的组的一个或更多个序列:smad4、gnas、tp53、raf1、cdk4、tyro3、mapk1、stk11、ccnd1、hras、met、myc和nras。
[0017]
在某些实施方案中,遗传变异或真正遗传变异包括单核苷酸变异(snv)或插入或缺失(indel)。在某些实施方案中,遗传变异包括snv。在某些实施方案中,snv位于内含子-外显子边界处。在某些实施方案中,snv位于基因编码序列(cds)内。在某些实施方案中,遗传变异包括插入或缺失。
[0018]
在某些实施方案中,所述区域包含与基因内融合断点相邻的约2个、4个、6个、8个、10个、15个或20个核苷酸。在某些实施方案中,该区域距离融合断点少于约100个、50个、20个、15个、10个、8个、6个、4个、2个核苷酸。在某些实施方案中,一个或更多个检测到的比对错误的一部分基于在样品中检测到的比对错误具有的突变等位基因分数小于或等于对应
于样品中的基因内融合断点的基因内融合的突变等位基因分数被过滤掉。在某些实施方案中,一个或更多个检测到的比对错误的一部分基于所述基因融合读段包含不属于临床可操作(clinically actionable)变异的预定义集合的遗传变异被过滤掉。
[0019]
在某些实施方案中,样品是选自由以下组成的组体液样品:血液、血浆、血清、尿液、唾液、粘膜分泌物、痰、粪便和泪液。在某些实施方案中,受试者患有疾病或紊乱。在某些实施方案中,疾病是癌症。
[0020]
在某些实施方案中,方法包括从受试者的生物样品分离无细胞核酸分子。在某些实施方案中,无细胞核酸分子包括dna、rna或它们的组合。在某些实施方案中,无细胞核酸分子是无细胞dna。在某些实施方案中,无细胞核酸分子是双链dna。
[0021]
在某些实施方案中,方法包括将一个或更多个包含分子条形码的衔接子附接至无细胞核酸分子,然后测序以产生加标签的亲本多核苷酸。在某些实施方案中,衔接子被附接至无细胞核酸分子的两端。在某些实施方案中,无细胞核酸分子被独特地加条形码。在某些实施方案中,无细胞核酸分子被非独特地加条形码。在某些实施方案中,每个条形码包含固定的或半随机的寡核苷酸序列,所述寡核苷酸序列与从选定区域测序的多种分子相结合,使得能够鉴定独特的分子。
[0022]
在某些实施方案中,方法包括扩增加标签的亲本多核苷酸以产生后代多核苷酸。在某些实施方案中,方法包括针对感兴趣的靶序列选择性富集后代多核苷酸,从而产生富集的后代多核苷酸。在某些实施方案中,方法包括扩增富集的后代多核苷酸。在某些实施方案中,方法包括用样品索引序列标记后代多核苷酸或富集的后代多核苷酸。
[0023]
在某些实施方案中,序列信息从核酸测序仪获得。在某些实施方案中,基因融合读段的集合通过将测序的成对末端读段进行比对和连接来鉴定。在某些实施方案中,基因融合读段的集合基于跨内含子-外显子边界的覆盖的不连续性来鉴定。在某些实施方案中,预定义集合包括存在于cosmic、癌症基因组图谱(the cancer genome atlas,cga)或外显子组聚合联盟(exome aggregation consortium,exac)中的变异。
[0024]
在某些实施例中,本发明的方法可以是计算机实现的,使得本说明书或所附权利要求书中描述的除了湿化学步骤之外的任何或所有步骤可以在合适的编程计算机中执行。
[0025]
在某些方面,本公开内容提供了一种系统,该系统包括控制器,该控制器包括或能够访问包括非暂时性计算机可执行指令的计算机可读介质,该指令在由至少一个电子处理器执行时,执行用于检测基因序列读段中的比对错误的方法,该方法包括:接收序列信息,所述序列信息包括从来自受试者的生物样品中的无细胞核酸分子获得的基因序列读段;将基因序列读段与参考序列比对以产生比对的序列读段;从比对的序列读段中鉴定包含基因内融合断点的基因融合读段的集合;以及通过鉴定在包含基因内融合断点的区域内包含遗传变异的基因融合读段的一个或更多个的子集来检测比对错误,其中该区域包含与基因内融合断点相邻的一个或更多个核苷酸。
[0026]
在某些方面,本公开内容提供了一种系统,该系统包括控制器,该控制器包括或能够访问包括非暂时性计算机可执行指令的计算机可读介质,该指令在由至少一个电子处理器执行时,执行用于在检测来自受试者的生物样品的无细胞核酸分子中的真正遗传变异时抑制比对错误的方法,该方法包括:接收序列信息,所述序列信息包括从无细胞核酸分子获得的序列读段;将序列读段与参考序列比对以产生比对的序列读段;从比对的序列读段中
鉴定包含基因内融合断点的基因融合读段的集合;通过鉴定在包含基因内融合断点的区域内包含遗传变异的基因融合读段的一个或更多个的子集来检测比对错误,其中该区域包含与基因内融合断点相邻的一个或更多个核苷酸;过滤掉一个或更多个基因融合读段的子集中的一个或更多个检测到的比对错误的至少一部分,以产生过滤的序列读段;以及检测与参考序列相比包含真正遗传变异的过滤的序列读段。
[0027]
在某些方面,本公开内容提供了一种系统,该系统包括控制器,该控制器包括或能够访问包括非暂时性计算机可执行指令的计算机可读介质,该指令在由至少一个电子处理器执行时,执行用于在检测来自受试者的样品的无细胞核酸分子中的真正遗传变异时抑制比对错误的方法,该方法包括:接收序列信息,所述序列信息包括从无细胞核酸分子获得的测序读段;将序列读段与参考序列比对以产生比对的序列读段;从比对的序列读段中鉴定包含基因内融合断点的基因融合读段的集合;通过鉴定包含遗传变异的基因融合读段的一个或更多个的子集来检测比对错误,其中该基因融合读段的一个或更多个的子集包括对应于smad4、tyro3和/或raf1的基因序列;过滤掉基因融合读段的一个或更多个的子集中的一个或更多个检测到的比对错误的至少一部分,以产生过滤的序列读段;以及检测与所述参考序列相比包含真正遗传变异的过滤的序列读段。
[0028]
在某些方面,本公开内容提供了一种系统,该系统包括控制器,该控制器包括或能够访问包括非暂时性计算机可执行指令的计算机可读介质,该指令在由至少一个电子处理器执行时,执行用于检测基因序列读段中的比对错误的方法,该方法包括:接收序列信息,所述序列信息包括从来自受试者的生物样品中的无细胞核酸分子获得的基因序列读段;将基因序列读段与参考序列比对以产生比对的序列读段;从比对的序列读段中确定包含基因内融合断点的基因融合读段的集合;确定在包含基因内融合断点的区域内包含遗传变异的基因融合读段的一个或更多个的子集,其中该区域包含与基因内融合断点相邻的一个或更多个核苷酸;以及将该区域内满足预定标准的每个遗传变异鉴定为比对错误。
[0029]
在某些方面,本公开内容提供了一种系统,该系统包括控制器,该控制器包括或能够访问包括非暂时性计算机可执行指令的计算机可读介质,该指令在由至少一个电子处理器执行时,执行用于在检测来自受试者的样品的无细胞核酸分子中的真正遗传变异时抑制比对错误的方法,该方法包括:接收序列信息,所述序列信息包括从来自受试者的生物样品中的无细胞核酸分子获得的序列读段;将序列读段与参考序列比对以产生比对的序列读段;从比对的序列读段中确定包含基因内融合断点的基因融合读段的集合;确定在包含基因内融合断点的区域内包含遗传变异的基因融合读段的一个或更多个的子集,其中该区域包含与基因内融合断点相邻的一个或更多个核苷酸;将该区域内满足预定标准的每个遗传变异鉴定为比对错误;过滤掉一个或更多个基因融合读段的子集中的一个或更多个比对错误,以产生过滤的序列读段;以及检测与参考序列相比包含真正遗传变异的过滤的序列读段。
[0030]
在某些方面,本公开内容提供了一种至少部分地使用计算机来产生过滤的序列信息数据集的方法,该方法包括:(a)接收测试序列信息,该测试序列信息包括从获自受试者的生物样品中的cfdna获得的测试序列读段;(b)鉴定测试序列读段中的一个或更多个分裂序列读段,其中每个分裂序列读段包含至少一个断点;以及(c)在测试序列信息中,抑制包含距离给定断点选定数目的核苷酸内的至少一个序列变异的一个或更多个分裂序列读段
的至少一部分和/或一个或更多个测试序列读段的至少一部分,从而产生过滤的序列信息数据集。
[0031]
在某些方面,本公开内容提供了一种至少部分地使用计算机来产生过滤的序列信息数据集的方法,该方法包括:(a)鉴定从获自受试者的生物样品中的cfdna获得的测试序列读段的集合中的一个或更多个分裂序列读段,其中每个分裂序列读段包含至少一个断点;以及(b)在测试序列读段的集合中,抑制包含距离给定断点选定数目的核苷酸内的至少一个序列变异的一个或更多个分裂序列读段的至少一部分和/或一个或更多个测试序列读段的至少一部分,从而产生过滤的序列信息数据集。
[0032]
在某些方面,本公开内容提供了一种至少部分地使用计算机来产生过滤的序列信息数据集的方法,该方法包括:(a)鉴定从获自受试者的生物样品中的cfdna获得的测试序列读段的集合中的一个或更多个分裂序列读段,其中每个分裂序列读段包含至少一个断点;以及(b)在测试序列读段的集合中,抑制包含距离给定断点选定数目的核苷酸内的至少一个序列变异的分裂序列读段的一个或更多个碱基识别和/或测试序列读段的一个或更多个碱基识别,从而产生过滤的序列信息数据集。
[0033]
在某些方面,本公开内容提供了一种至少部分地使用计算机来产生过滤的序列信息数据集的方法,该方法包括:(a)接收测试序列信息,该测试序列信息包括从获自受试者的生物样品中的cfdna获得的测试序列读段;(b)鉴定测试序列读段中的一个或更多个分裂序列读段,其中每个分裂序列读段包含至少一个断点;以及(c)在测试序列信息中,抑制包含距离给定断点选定数目的核苷酸内的至少一个序列变异的分裂序列读段的一个或更多个碱基识别和/或测试序列读段的一个或更多个碱基识别,从而产生过滤的序列信息数据集。
[0034]
在某些方面,本公开内容提供了一种至少部分地使用计算机来产生过滤的序列信息数据集的方法,该方法包括:(a)鉴定从获自受试者的生物样品中的无细胞核酸(cfna)获得的测试序列读段的集合中的一个或更多个分裂序列读段,其中每个分裂序列读段包含至少一个断点;以及(b)在测试序列读段的集合中,抑制包含距离给定断点选定数目的核苷酸内的至少一个序列变异的一个或更多个分裂序列读段的至少一部分和/或一个或更多个测试序列读段的至少一部分,从而产生过滤的序列信息数据集。
[0035]
在某些方面,本公开内容提供了一种至少部分地使用计算机来产生过滤的序列信息数据集的方法,该方法包括:(a)鉴定从获自受试者的生物样品中的无细胞核酸(cfna)获得的测试序列读段的集合中的一个或更多个分裂序列读段,其中每个分裂序列读段包含至少一个断点;以及(b)在测试序列读段的集合中,抑制包含距离给定断点选定数目的核苷酸内的至少一个序列变异的分裂序列读段的一个或更多个碱基识别和/或测试序列读段的一个或更多个碱基识别,从而产生过滤的序列信息数据集。
[0036]
在某些方面,本公开内容提供了一种产生过滤的序列信息数据集的方法,该方法包括:(a)对从受试者获得的生物样品中的无细胞脱氧核糖核酸(cfdna)进行测序以产生测试序列读段的集合;(b)鉴定测试序列读段的集合中的一个或更多个分裂序列读段,其中每个分裂序列读段包含至少一个断点;以及(c)在测试序列读段的集合中,抑制包含距离给定断点选定数目的核苷酸内的至少一个序列变异的一个或更多个分裂序列读段的至少一部分和/或一个或更多个测试序列读段的至少一部分,从而产生过滤的序列信息数据集。
[0037]
在某些方面,本公开内容提供了一种至少部分地使用计算机来检测靶序列变异的方法,该方法包括:(a)鉴定从获自受试者的生物样品中的cfdna获得的测试序列读段的集合中的一个或更多个分裂序列读段,其中每个分裂序列读段包含至少一个断点;(b)在测试序列读段的集合中,抑制包含距离给定断点选定数目的核苷酸内的至少一个非靶序列变异的一个或更多个分裂序列读段的至少一部分和/或一个或更多个测试序列读段的至少一部分,以产生过滤的序列信息数据集;以及(c)鉴定包含靶序列变异的过滤的序列信息数据集中的至少一个靶测试序列读段,从而检测靶序列变异。
[0038]
在某些方面,本公开内容提供了一种治疗受试者的疾病、紊乱或状况的方法,该方法包括:(a)鉴定从获自受试者的生物样品中的cfdna获得的测试序列读段的集合中的一个或更多个分裂序列读段,其中每个分裂序列读段包含至少一个断点;(b)在测试序列读段的集合中,抑制包含距离给定断点选定数目的核苷酸内的至少一个非靶序列变异的一个或更多个分裂序列读段的至少一部分和/或一个或更多个测试序列读段的至少一部分,以产生过滤的序列信息数据集;(c)鉴定包含靶序列变异的过滤的序列信息数据集中的至少一个靶测试序列读段,该靶序列变异指示受试者的疾病、紊乱或状况;以及(d)向受试者施用在治疗疾病、紊乱或状况方面有效的一种或更多种疗法,从而治疗受试者的疾病、紊乱或状况。
[0039]
在某些实施方案中,方法包括当另外的测试序列读段与选自由以下组成的组的一个或更多个基因序列的至少一部分对齐(align)时,抑制包含不在距离给定断点选定数目的核苷酸内的一个或更多个序列变异的一个或更多个另外的测试序列读段:smad4、gnas、tp53、raf1、cdk4、tyro3、mapk1、stk11、ccnd1、hras、met、myc和nras。
[0040]
在某些实施方案中,鉴定给定的分裂序列读段包括鉴定仅与参考序列信息部分对齐的测试序列读段。在某些实施方案中,鉴定给定的分裂序列读段包括鉴定测试序列信息中一个或更多个基因组区域相对于参考序列信息增加的覆盖,所述参考序列信息缺乏包含一个或更多个基因组区域的分裂序列读段。
[0041]
在某些实施方案中,一个或更多个基因组区域包括至少一个编码序列(cds)。在某些实施例中,鉴定给定的分裂序列读段包括鉴定至少两个彼此不同并且各自包含相同断点的分裂序列读段。在某些实施例中,方法包括鉴定过滤的序列信息数据集中的至少一个靶测试序列读段。在某些实施方案中,靶测试序列读段包含指示受试者的给定疾病、紊乱或状况的靶序列变异。在某些实施方案中,方法包括治疗受试者的给定疾病、紊乱或状况。
[0042]
在某些实施方案中,一个或更多个被抑制的分裂序列读段包含加工的假基因(ppg)的至少一部分。在某些实施方案中,方法包括从测试序列信息中去除包含距离给定断点选定数目的核苷酸内的序列变异的分裂序列读段和/或测试序列读段。
[0043]
在某些方面,本公开内容提供了一种系统,该系统包括控制器,该控制器包括或能够访问包括非暂时性计算机可执行指令的计算机可读介质,当该指令被至少一个电子处理器执行时执行至少以下操作:(a)接收测试序列信息,该测试序列信息包括从获自受试者的生物样品中的cfdna获得的测试序列读段;(b)鉴定测试序列读段中的一个或更多个分裂序列读段,其中每个分裂序列读段包含至少一个断点;以及(c)在测试序列信息中,抑制包含距离给定断点选定数目的核苷酸内的至少一个序列变异的一个或更多个分裂序列读段的至少一部分和/或一个或更多个测试序列读段的至少一部分。
[0044]
在某些方面,本公开内容提供了一种系统,该系统包括控制器,该控制器包括或能够访问包括非暂时性计算机可执行指令的计算机可读介质,当该指令被至少一个电子处理器执行时执行至少以下操作:(a)鉴定从获自受试者的生物样品中的cfdna获得的测试序列读段的集合中的一个或更多个分裂序列读段,其中每个分裂序列读段包含至少一个断点;以及(b)在测试序列读段的集合中,抑制包含距离给定断点选定数目的核苷酸内的至少一个序列变异的一个或更多个分裂序列读段的至少一部分和/或一个或更多个测试序列读段的至少一部分。
[0045]
在某些方面,本公开内容提供了一种系统,该系统包括控制器,该控制器包括或能够访问包括非暂时性计算机可执行指令的计算机可读介质,当该指令被至少一个电子处理器执行时执行至少以下操作:(a)鉴定从获自受试者的生物样品中的cfdna获得的测试序列读段的集合中的一个或更多个分裂序列读段,其中每个分裂序列读段包含至少一个断点;以及(b)在测试序列读段的集合中,抑制包含距离给定断点选定数目的核苷酸内的至少一个序列变异的分裂序列读段的一个或更多个碱基识别和/或测试序列读段的一个或更多个碱基识别。
[0046]
在某些方面,本公开内容提供了一种系统,该系统包括控制器,该控制器包括或能够访问包括非暂时性计算机可执行指令的计算机可读介质,当该指令被至少一个电子处理器执行时执行至少以下操作:(a)接收测试序列信息,该测试序列信息包括从获自受试者的生物样品中的cfdna获得的测试序列读段;(b)鉴定测试序列读段中的一个或更多个分裂序列读段,其中每个分裂序列读段包含至少一个断点;以及(c)在测试序列信息中,抑制包含距离给定断点选定数目的核苷酸内的至少一个序列变异的分裂序列读段的一个或更多个碱基识别和/或测试序列读段的一个或更多个碱基识别。
[0047]
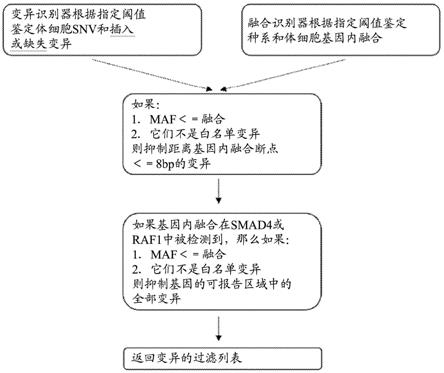
在某些方面,本公开内容提供了一种系统,该系统包括控制器,该控制器包括或能够访问包括非暂时性计算机可执行指令的计算机可读介质,当该指令被至少一个电子处理器执行时执行至少以下操作:(a)鉴定从获自受试者的生物样品中的无细胞核酸(cfna)获得的测试序列读段的集合中的一个或更多个分裂序列读段,其中每个分裂序列读段包含至少一个断点;以及(b)在测试序列读段的集合中,抑制包含距离给定断点选定数目的核苷酸内的至少一个序列变异的一个或更多个分裂序列读段的至少一部分和/或一个或更多个测试序列读段的至少一部分。
[0048]
在某些方面,本公开内容提供了一种系统,该系统包括控制器,该控制器包括或能够访问包括非暂时性计算机可执行指令的计算机可读介质,当该指令被至少一个电子处理器执行时执行至少以下操作:(a)鉴定从获自受试者的生物样品中的cfna获得的测试序列读段的集合中的一个或更多个分裂序列读段,其中每个分裂序列读段包含至少一个断点;以及(b)在测试序列读段的集合中,抑制包含距离给定断点选定数目的核苷酸内的至少一个序列变异的分裂序列读段的一个或更多个碱基识别和/或测试序列读段的一个或更多个碱基识别。
[0049]
在某些方面,本公开内容提供了一种系统,该系统包括控制器,该控制器包括或能够访问包括非暂时性计算机可执行指令的计算机可读介质,当该指令被至少一个电子处理器执行时执行至少以下操作:(a)对从受试者获得的生物样品中的cfdna进行测序以产生测试序列读段的集合;(b)鉴定测试序列读段的集合中的一个或更多个分裂序列读段,其中每
个分裂序列读段包含至少一个断点;以及(c)在测试序列读段的集合中,抑制包含距离给定断点选定数目的核苷酸内的至少一个序列变异的一个或更多个分裂序列读段的至少一部分和/或一个或更多个测试序列读段的至少一部分。
[0050]
在某些方面,本公开内容提供了一种系统,该系统包括控制器,该控制器包括或能够访问包括非暂时性计算机可执行指令的计算机可读介质,当该指令被至少一个电子处理器执行时执行至少以下操作:(a)鉴定从获自受试者的生物样品中的cfdna获得的测试序列读段的集合中的一个或更多个分裂序列读段,其中每个分裂序列读段包含至少一个断点;(b)在测试序列读段的集合中,抑制包含距离给定断点选定数目的核苷酸内的至少一个非靶序列变异的一个或更多个分裂序列读段的至少一部分和/或一个或更多个测试序列读段的至少一部分,以产生过滤的序列信息数据集;以及(c)鉴定包含靶序列变异的过滤的序列信息数据集中的至少一个靶测试序列读段。
[0051]
在某些方面,本公开内容提供了一种包括非暂时性计算机可执行指令的计算机可读介质,当该指令被至少一个电子处理器执行时执行至少以下操作:(a)接收测试序列信息,该测试序列信息包括从获自受试者的生物样品中的无细胞脱氧核糖核酸(cfdna)获得的测试序列读段;(b)鉴定测试序列读段中的一个或更多个分裂序列读段,其中每个分裂序列读段包含至少一个断点;以及(c)在测试序列信息中,抑制包含距离给定断点选定数目的核苷酸内的至少一个序列变异的一个或更多个分裂序列读段的至少一部分和/或一个或更多个测试序列读段的至少一部分。
[0052]
在某些方面,本公开内容提供了一种包括非暂时性计算机可执行指令的计算机可读介质,当该指令被至少一个电子处理器执行时执行至少以下操作:(a)鉴定从获自受试者的生物样品中的cfdna获得的测试序列读段的集合中的一个或更多个分裂序列读段,其中每个分裂序列读段包含至少一个断点;以及(b)在测试序列读段的集合中,抑制包含距离给定断点选定数目的核苷酸内的至少一个序列变异的一个或更多个分裂序列读段的至少一部分和/或一个或更多个测试序列读段的至少一部分。
[0053]
在某些方面,本公开内容提供了一种包括非暂时性计算机可执行指令的计算机可读介质,当该指令被至少一个电子处理器执行时执行至少以下操作:(a)鉴定从获自受试者的生物样品中的cfdna获得的测试序列读段的集合中的一个或更多个分裂序列读段,其中每个分裂序列读段包含至少一个断点;以及(b)在测试序列读段的集合中,抑制包含距离给定断点选定数目的核苷酸内的至少一个序列变异的分裂序列读段的一个或更多个碱基识别和/或测试序列读段的一个或更多个碱基识别。
[0054]
在某些方面,本公开内容提供了一种包括非暂时性计算机可执行指令的计算机可读介质,当该指令被至少一个电子处理器执行时执行至少以下操作:(a)接收测试序列信息,该测试序列信息包括从获自受试者的生物样品中的cfdna获得的测试序列读段;(b)鉴定测试序列读段中的一个或更多个分裂序列读段,其中每个分裂序列读段包含至少一个断点;以及(c)在测试序列信息中,抑制包含距离给定断点选定数目的核苷酸内的至少一个序列变异的分裂序列读段的一个或更多个碱基识别和/或测试序列读段的一个或更多个碱基识别。
[0055]
在某些方面,本公开内容提供了一种包括非暂时性计算机可执行指令的计算机可读介质,当该指令被至少一个电子处理器执行时执行至少以下操作:(a)鉴定从获自受试者
的生物样品中的cfna获得的测试序列读段的集合中的一个或更多个分裂序列读段,其中每个分裂序列读段包含至少一个断点;以及(b)在测试序列读段的集合中,抑制包含距离给定断点选定数目的核苷酸内的至少一个序列变异的一个或更多个分裂序列读段的至少一部分和/或一个或更多个测试序列读段的至少一部分。
[0056]
在某些方面,本公开内容提供了一种包括非暂时性计算机可执行指令的计算机可读介质,当该指令被至少一个电子处理器执行时执行至少以下操作:(a)鉴定从获自受试者的生物样品中的cfna获得的测试序列读段的集合中的一个或更多个分裂序列读段,其中每个分裂序列读段包含至少一个断点;以及(b)在测试序列读段的集合中,抑制包含距离给定断点选定数目的核苷酸内的至少一个序列变异的分裂序列读段的一个或更多个碱基识别和/或测试序列读段的一个或更多个碱基识别。
[0057]
在某些方面,本公开内容提供了一种包括非暂时性计算机可执行指令的计算机可读介质,当该指令被至少一个电子处理器执行时执行至少以下操作:(a)对从受试者获得的生物样品中的无细胞脱氧核糖核酸(cfdna)进行测序以产生测试序列读段的集合;(b)鉴定测试序列读段的集合中的一个或更多个分裂序列读段,其中每个分裂序列读段包含至少一个断点;以及(c)在测试序列读段的集合中,抑制包含距离给定断点选定数目的核苷酸内的至少一个序列变异的一个或更多个分裂序列读段的至少一部分和/或一个或更多个测试序列读段的至少一部分。
[0058]
在某些方面,本公开内容提供了一种包括非暂时性计算机可执行指令的计算机可读介质,当该指令被至少一个电子处理器执行时执行至少以下操作:(a)鉴定从获自受试者的生物样品中的cfdna获得的测试序列读段的集合中的一个或更多个分裂序列读段,其中每个分裂序列读段包含至少一个断点;(b)在测试序列读段的集合中,抑制包含距离给定断点选定数目的核苷酸内的至少一个非靶序列变异的一个或更多个分裂序列读段的至少一部分和/或一个或更多个测试序列读段的至少一部分,以产生过滤的序列信息数据集;以及(c)鉴定包含靶序列变异的过滤的序列信息数据集中的至少一个靶测试序列读段。
[0059]
附图简述
[0060]
图1是示出用于检测和抑制由于加工的假基因的存在而导致的比对错误的示例性方法的图示。
[0061]
图2是示意性描绘根据本公开内容的一些实施方案产生过滤的序列信息数据集的示例性方法步骤的流程图。
[0062]
图3是示意性描绘根据本公开内容的一些实施方案产生过滤的序列信息数据集的示例性方法步骤的流程图。
[0063]
图4a是示出藉以产生加工的假基因的过程的图示。人类line元件中存在的非特异性逆转录酶机制产生加工的(即,无内含子)mrna的dna拷贝并将其整合到基因组中。
[0064]
图4b是示出源自假基因的读段可以如何独特地映射(map)到原始基因的图示,因为样品特异性ppg不在人类基因组组装(human genome assembly)(例如,hg19)中。然而,假基因的存在可以通过源自跨内含子-外显子边界的ppg片段的分裂读段的存在来揭示。
[0065]
图5是示出了被编程或以其他方式配置成实现本文提供的方法的计算机系统的图示。
[0066]
图6是示出映射到smad4外显子11的序列读段的图示。源自单个分子的读段按颜色
(即灰阶(greyscale)阴影)和共同的基因组坐标进行分组。缺少内含子序列的多个软剪读段(soft-clipped read)(读段右手侧的多色图案)的存在以及内含子-外显子边界处覆盖的不连续性(图的顶部)揭示了ppg的存在。以1.7%的等位基因频率观察到箭头指示的假的a>c snv识别。
[0067]
图7a是示出当检测到ppg时,在hras、smad4和pt53中以高于不含ppg的样品中本来预期的比率观察到剪接接点(junctions)中的snv识别的图。在10,000个随机背景样品中,在这些相同的接点内没有识别snv,并且由此灰色背景条处于相同的高度0,且因此不可见。
[0068]
图7b是示出当检测到ppg时,在smad4和raf1的编码序列(cds)内snv以更高比率被识别的图。示出所有具有>=含有ppg的样品的基因;当ppg存在时,gnas和tp53都没有显示出更高比率的cds snv识别。基于卡方检验(1d.f.),***,p<0.01,*p<0.05;n.s.,不显著。
[0069]
图8是示出映射到人类染色体15上的tyro3的序列读段的图示。源自单个分子的读段按颜色(即灰阶阴影)和共同的基因组坐标进行分组。由ppg产生的跨外显子-外显子接点的比对假象显示在tyro3基因座的背景中。假的c.t.snv识别(tyro3 c.1422c>t)由箭头指示。
[0070]
定义
[0071]
术语“受试者”可以指动物,诸如哺乳动物物种(优选人类)或禽类(例如鸟类)物种。更具体地,受试者可以是脊椎动物、例如哺乳动物,诸如,小鼠、灵长类动物、猿猴或人类。动物包括农场动物、运动动物和宠物。受试者可以是健康个体、具有症状或病征或怀疑患有疾病或易患该疾病的个体,或者需要治疗或怀疑需要治疗的个体。在一些实施方案中,受试者是人类,诸如患有癌症或被怀疑患有癌症的人。
[0072]
措辞“无细胞核酸”可以指不包含在细胞内或本来不与细胞结合的核酸,或者换言之,在去除完整细胞后保留在样品中的核酸。无细胞核酸可以指来源于受试者的体液(例如,血液、尿液、csf等)的非包封的核酸。无细胞核酸包括dna(cfdna)、rna(cfrna)及其杂交体,包括基因组dna、线粒体dna、循环dna、sirna、mirna、循环rna(crna)、trna、rrna、小核仁rna(snorna)、piwi-相互作用rna(pirna)、长非编码rna(长ncrna)或这些中任何一种的片段。无细胞核酸可以是双链的、单链的或部分双链和单链的。无细胞核酸可以通过分泌或细胞死亡过程,例如细胞坏死和凋亡,释放到体液中。一些无细胞核酸是从癌细胞释放到体液中的,例如,循环肿瘤dna(ctdna)。其他的是从健康细胞释放的。ctdna可以是非包封的肿瘤来源的片段化dna。无细胞胎儿dna(cffdna)是在母体血流中自由循环的胎儿dna。无细胞核酸可以具有一种或更多种相关的表观遗传修饰,例如,可以被乙酰化、5-甲基化、泛素化、磷酸化、类泛素化(sumoylated)、核糖基化和/或瓜氨酸化(citrullinated)。在一些实施方案中,无细胞核酸是通常包括双链cfdna的cfdna。
[0073]
措辞“核酸标签”可以指短核酸(例如,长度小于500个、100个、50个或10个核苷酸),用于标记核酸分子以区分来自不同样品(例如,呈现为样品索引(sample index))的核酸,或同一样品中不同类型的或经历不同处理的不同核酸分子(例如,呈现为分子条形码)。标签可以是单链、双链或至少部分双链的。标签可以具有相同的长度或不同的长度。标签可以是平端的,或者具有突出端。标签可以附接至核酸的一端或两端。核酸标签可以被解码以揭示诸如核酸的样品来源、形式或加工的信息。标签可以用于允许汇集和并行处理包含带有不同分子标签和/或样品索引的核酸的多个样品,随后通过读取分子标签对核酸进行解
卷积。另外或可选地,核酸标签可以用于区分同一样品中的不同分子(即,分子条形码)。这既包括对样品中的不同分子独特地加标签,也包括对样品中的分子非独特地加标签。在非独特加标签的情况下,可以使用有限数目的不同标签来标记分子,使得可以基于不同分子映射在参考基因组上的其起始和/或终止位置(即,基因组坐标)以及至少一个标签来区分不同分子。通常则使用足够数目的不同标签,使得具有相同起始/终止的任何两个分子也具有相同标签的概率较低(例如<10%、<5%、<1%或<0.1%)。一些标签包含多个标识符来标记样品、样品内的分子形式以及具有相同起始点和终止点的形式内的分子。这样的标签可以以a1i的形式存在,其中字母指示样品类型,阿拉伯数字指示样品内的分子形式,而罗马数字指示形式内的分子。
[0074]
术语“衔接子”是指通常至少部分双链的短核酸(例如,长度小于500个、100个或50个核苷酸),用于连接至样品核酸分子的任一端或两端。衔接子可以包含允许扩增两端侧接衔接子的核酸分子的引物结合位点,和/或测序引物结合位点,包括用于下一代测序(ngs)的引物结合位点。衔接子也可以包含用于捕获探针的结合位点,诸如附接至流动池支持物的寡核苷酸。衔接子也可以包含如上文描述的标签。标签优选地相对于引物结合位点和测序引物结合位点来放置,使得标记被包含在核酸分子的扩增子和测序读段中。相同序列或不同序列的衔接子可以被连接至核酸分子的相应端。有时除了条形码不同的相同序列的衔接子被连接至相应端。优选的衔接子是y形衔接子,其中一端是平端或加尾的,用于连接核酸分子,核酸分子也是平端或加尾的或用一个或更多个互补核苷酸加尾的。另一种优选的衔接子是钟形衔接子,同样具有用于连接待分析核酸的平端或加尾端。
[0075]
如本文使用的,术语“测序”或“测序仪”是指用于确定生物分子(例如核酸,诸如dna或rna)序列的多种技术中的任何一种。示例性测序方法包括但不限于靶向测序、单分子实时测序、外显子测序、基于电子显微术的测序、panel测序、晶体管介导的测序、直接测序、随机鸟枪法测序、sanger双脱氧终止测序、全基因组测序、杂交测序、焦磷酸测序、双链体测序、循环测序、单碱基延伸测序、固相测序、高通量测序、大规模并行信号测序(massively parallel signature sequencing)、乳液pcr、低变性温度共扩增pcr(cold-pcr)、多重pcr、可逆染料终止子测序、配对末端测序、near-term测序、外切核酸酶测序、连接测序、短读段测序、单分子测序、合成测序、实时测序、反向终止子测序、纳米孔测序、454测序、solexa基因组分析仪测序、solid
tm
测序、ms-pet测序及其组合。在一些实施方案中,测序可以通过诸如例如从illumina或applied biosystems商业可获得的基因分析仪进行。
[0076]
措辞“下一代测序”或ngs是指与传统的基于sanger和毛细管电泳的方法相比具有增加的通量的测序技术,例如,具有一次产生数十万个相对小的序列读段的能力。下一代测序技术的一些实例包括但不限于合成测序、连接测序和杂交测序。
[0077]
术语“dna(脱氧核糖核酸)”是指包含脱氧核糖核苷的核苷酸链,每个脱氧核糖核苷包括四个核酸碱基之一,即腺嘌呤(a)、胸腺嘧啶(t)、胞嘧啶(c)和鸟嘌呤(g)。术语“rna(核糖核酸)”是指包括四种类型的核糖核苷的核苷酸链,每种核糖核苷包括四种核酸碱基之一,即:a、尿嘧啶(u)、g和c。某些核苷酸对以互补方式彼此特异性结合(称为互补碱基对)。在dna中,腺嘌呤(a)与胸腺嘧啶(t)配对,胞嘧啶(c)与鸟嘌呤(g)配对。在rna中,腺嘌呤(a)与尿嘧啶(u)配对,胞嘧啶(c)与鸟嘌呤(g)配对。当第一条核酸链与由与第一条链的核苷酸互补的核苷酸构成的第二条核酸链结合时,两条链结合形成双链。如本文使用的,
“
核酸测序数据”、“核酸测序信息”、“核酸序列”、“核苷酸序列”、“基因组序列”、“基因序列”或“片段序列”或“核酸测序读段”表示指示核酸(诸如,dna或rna)的分子(例如,全基因组、全转录组、外显子组、寡核苷酸、多核苷酸或片段)中核苷酸碱基(例如,腺嘌呤、鸟嘌呤、胞嘧啶和胸腺嘧啶或尿嘧啶)顺序的任何信息或数据。应当理解,本教导设想了使用所有可用的各种技术(technique)、平台或技术(technology)获得的序列信息,包括但不限于:毛细管电泳、微阵列、基于连接的系统、基于聚合酶的系统、基于杂交的系统、直接或间接核苷酸鉴定系统、焦磷酸测序、基于离子或ph的检测系统以及基于电子信号的系统。
[0078]“多核苷酸”、“核酸”、“核酸分子”或“寡核苷酸”是指通过核苷间键连接的核苷(包括脱氧核糖核苷、核糖核苷或其类似物)的线性聚合物。通常,多核苷酸包含至少三个核苷。寡核苷酸的尺寸范围通常从几个单体单元(例如,3-4个)到数百个单体单元。每当多核苷酸以一串字母诸如“atgcctg”表示时,应当理解,这些核苷酸从左到右为5
’→3’
顺序,并且“a”表示腺苷,“c”表示胞嘧啶,“g”表示鸟苷,并且“t”表示胸苷,除非另外说明。字母a、c、g和t可以用来指碱基本身、包含碱基的核苷或核苷酸,这是本领域的标准。
[0079]“参考序列”是指用于与实验确定的序列进行比较的目的的已知序列。例如,已知序列可以是整个基因组、染色体或其任何区段。参考通常包括至少20个、50个、100个、200个、250个、300个、350个、400个、450个、500个、1000个或更多个核苷酸。参考序列可以与基因组或染色体的单个连续序列对齐,或者可以包括与基因组或染色体的不同区域对齐的非连续区段。在一些实施方案中,参考序列是人类基因组。参考人类基因组包括例如hg19和hg38。
[0080]
术语“假基因”通常是指基因序列与对应的完整基因相似,但在细胞基因表达或蛋白编码能力方面丧失了至少一些功能的基因组dna的区段。假基因可能与其功能对应基因具有高度同源性或同一性。在一些实施方案中,假基因与对应的功能基因共有至少40%、至少45%、至少50%、至少55%、至少60%、至少65%、至少70%、至少75%、至少80%、至少85%、至少90%或至少95%的同源性。
[0081]
措辞“加工的假基因”通常是指由逆转录转座过程产生的假基因,由此互补dna(cdna)(逆转录的mrna转录物)重新整合到基因组的新位置中。加工的假基因通常缺少内含子,从而产生外显子-外显子的基因内(即,基因之内)融合。加工的假基因的其他特征包括多聚a尾、截短的5’端(与对应的完整基因相比)且缺少转录机制(例如启动子区域)。
[0082]
如本文使用的措辞“生物样品”通常指源自受试者的组织或流体样品。生物样品可以直接从受试者获得。生物样品可以是一种或更多种核酸分子或者可以包括一种或更多种核酸分子,诸如脱氧核糖核酸(dna)分子或核糖核酸(rna)分子。生物样品可以源自任何器官、组织或生物流体。生物样品可以包括例如体液或固体组织样品。固体组织样品的实例是肿瘤样品,例如来自实体瘤活组织检查的样品。体液包括例如血液、血清、血浆、肿瘤细胞、唾液、尿液、淋巴液、前列腺液、精液、乳汁、痰、粪便、泪液及它们的衍生物。在一些实施方案中,生物样品是血液或源自血液。
[0083]
措辞“突变等位基因分数”、“突变剂量”或“maf”是指在给定样品的给定基因组位置处含有等位基因改变或突变的核酸分子的分数。maf通常表示为分数或百分比。例如,maf通常小于给定基因座处存在的所有体细胞变异或等位基因的约0.5、0.1、0.05或0.01%(即,小于约50%、10%、5%或1%)。
[0084]
在核酸序列信息的上下文中,措辞“分裂序列读段(split sequence read)”或“分裂读段(split read)”或“基因融合读段”是指包含映射到给定参考序列的不同的非连续区域或基因座的子序列的测序读段。在某些实施方案中,例如,给定分裂序列读段的第一子序列映射到参考序列的给定基因的第一外显子,而该给定分裂序列读段的第二子序列映射到参考序列的相同基因的第二外显子,其中第一外显子和第二外显子被参考序列的相同基因的中间内含子隔开。在这些实施方案中的一些中,这样的分裂序列读段指示受试者(从其获得给定分裂序列读段)的基因组中存在基因内融合。在其他示例性实施方案中,给定分裂序列读段的第一子序列映射到参考序列的第一基因的外显子,而该给定分裂序列读段的第二子序列映射到参考序列的不同的第二基因的外显子,这些外显子在参考序列中彼此不连续。在这些实施方案中的一些中,这样的分裂序列读段指示受试者(从其获得给定分裂序列读段)的基因组中存在基因间融合。
[0085]
术语“断点”在核酸融合分子或相应的测序读段的上下文中是指在核酸融合的融合子序列之间的接点处或在相应的测序读段中呈现的末端核苷酸位置。例如,给定分裂序列读段可以包含与该分裂序列读段中的第二子序列相邻并且相对于第二子序列在5’的第一子序列,其中第一子序列映射到参考序列中的第一基因座,该第一基因座与第二子序列映射到该参考序列中的第二基因座不连续。在该实例中,分裂序列读段的第一子序列在其3’末端核苷酸处包含断点,而分裂序列读段的第二子序列在其5’末端核苷酸处包含断点。在某些应用中,诸如这些的断点被称为“断点对”。
[0086]
在治疗剂(例如,治疗性核酸构建体)的上下文中,措辞“施用”意指向受试者给予、施加剂或使剂与受试者接触。施用可以通过多种途径中的任何一种来完成,包括例如局部施用、口服施用、皮下施用、肌内施用、腹膜内施用、静脉内施用、鞘内施用和皮内施用。
[0087]
如用于一个或更多个感兴趣的值或要素的措辞“约”或“近似地”是指与所述参考值或要素相似的值或要素。在某些实施方案中,术语“约”或“近似地”是指在所述参考值或要素的任一方向上(大于或小于)落在25%、20%、19%、18%、17%、16%、15%、14%、13%、12%、11%、10%、9%、8%、7%、6%、5%、4%、3%、2%、1%或更小的值或要素的范围内,除非另有说明或另外从上下文中明显的(除非该数字超过可能的值或要素的100%)。
[0088]
详细描述
[0089]
i.总体概述
[0090]
对于临床诊断测序测试的一个核心挑战是鉴定容易出现短读假象(short-read artefact)的基因组区域并减轻其影响。这些区域中的许多已经通过人类基因组组装的分析而被鉴定出;然而,样品特异性融合事件(其中野生型染色体的总体结构被改变而使不相邻的基因组区域在同一条染色体上非常接近)或者逆转录假象(诸如由种系和体细胞的加工的假基因(ppg)的存在产生的假象)如果没有被正确鉴定出,则可以以体细胞等位基因频率产生假阳性变异识别。通过在逐个样品的基础上鉴定由这些融合事件产生的信号,本文公开的方法和系统可以鉴定和消除临床误导性变异的重要来源,同时以最小的灵敏度代价保持高特异性。
[0091]
本文提供的方法和系统在核酸分子、特别是无细胞核酸分子的分析中可能特别有用。在一些情况下,可以从来自受试者的生物样品提取并分离无细胞核酸分子。生物样品可以包括体液样品,体液样品选自包括但不限于以下的组:血液、血浆、血清、尿液、唾液、粘膜
分泌物、痰、粪便和泪液。无细胞核酸分子可以使用本领域中已知的多种方法进行提取,该多种方法包括但不限于异丙醇沉淀和/或硅基纯化。
[0092]
生物样品可以从多种受试者收集,诸如无疾病的受试者,处于诸如癌症或病毒的疾病的风险、表现出诸如癌症或病毒的疾病的症状或者患有诸如癌症或病毒的疾病的受试者,或处于遗传性紊乱的风险、表现出遗传性紊乱的症状或患有遗传性紊乱的受试者。在一些实施方案中,疾病或紊乱选自由以下组成的组:免疫缺陷紊乱、血友病、地中海贫血、镰状细胞病、血液病、慢性肉芽肿性紊乱、先天性盲、溶酶体贮积病、肌肉萎缩症、癌症、神经退行性疾病或它们的组合。在一些实施方案中,疾病是癌症。
[0093]
在获得或提供无细胞核酸分子后,可以对无细胞核酸分子进行用于制备用于测序的核酸分子的多种不同文库制备程序中的任何一种。无细胞核酸分子在测序之前可以用一种或更多种试剂(例如,酶、衔接子、标签(例如条形码)、探针等)进行处理。加标签的分子然后可以用于下游应用,诸如测序反应,藉此可以追溯个体分子。
[0094]
在一些实施方案中,方法还可以包括测序前的富集步骤,由此加标签的分子的区域被选择性或非选择性富集。
[0095]
在无细胞核酸分子的测序数据被收集后,一个或更多个生物信息学过程可以被应用于序列数据以检测比对错误(例如,假阳性序列读段),诸如由ppg的存在引起的比对错误,并且当提供遗传测序测试的结果时抑制或消除比对错误。此类过程可包括但不限于鉴定种系和体细胞基因融合序列读段,鉴定序列读段中的体细胞单核苷酸变异(snv)和/或插入或缺失(indel),确定基因融合断点(例如,基因内或基因间)的区域内的比对错误,应用过滤器以基于预定标准从序列读段或从检测到的变异的最终集合中去除比对错误,以及从过滤的序列读段中鉴定真正的遗传变异。
[0096]
在一些情况下,由测序反应产生的序列读段可以与参考序列进行比对,以进行生物信息学分析。在生物信息学分析的多个方面,可以设置一个或更多个阈值来确保质量。例如,可以设置比对阈值,使得只有高度相似的序列读段(例如,参考序列和序列读段之间有10个或更少的错配)被映射到参考序列。在一些情况下,可以去除不能通过质量阈值的序列读段,例如基于序列读段的色谱图(chromatogram)。在一些情况下,给定序列的拷贝数或量可以基于映射到给定序列或与给定序列对齐的序列读段的数目来定量。在一些情况下,序列的过度表现(over-representation)可以通过比较所有序列读段中不同序列的拷贝数或量来确定。
[0097]
在某些实施方案中,可以使样品与足够数量的衔接子接触,使得同一核酸的任何两个拷贝从一端或两端处连接的衔接子接收到相同的衔接子分子条形码组合的概率较低(例如,<1%)。以这种方式使用衔接子可以允许将具有相同起始点和终止点的序列读段分组为从同一原始分子产生的读段的家族,所述起始点和终止点与参考序列对齐(或映射到参考序列)并且连接至相同的条形码组合。这样的家族可以代表扩增前样品中核酸的扩增产物的序列。
[0098]
在一些实施方案中,可以对家族成员的序列进行汇编,以获得原始样品中的核酸分子的共有核苷酸或完整的共有序列,所述核酸分子通过平端形成和衔接子附接被修饰。换言之,占据样品中核酸的特定位置的核苷酸可以被确定为占据家族成员序列中对应位置的核苷酸的共有核苷酸。共有核苷酸可以通过诸如投票评分或置信评分的方法来确定,这
是两种非限制性的示例性方法。家族可以包括双链核酸的一条链或两条链的序列。如果家族的成员包括来自双链核酸的两条链的序列,为了对所有序列汇编以获得共有核苷酸或序列的目的,一条链的序列被转化为它们的互补序列。一些家族可能只包括单个成员序列。在这种情况下,该序列可以被视为扩增前样品中的核酸的序列。可选地,只有单个成员序列的家族可以从随后的分析中被排除。
[0099]
参考序列可以是一个或更多个已知序列,例如来自给定受试者的已知的全基因组序列或部分基因组序列,诸如人类受试者的全基因组序列。参考序列可以是hg19。如上所述,测序的核酸可以代表直接确定的样品中的核酸的序列,或者这种核酸的扩增产物的共有序列。可以在参考序列上的一个或更多个指定位置处进行比较。当对应序列被最大程度地对齐时,可以鉴定测序的核酸的子集,该子集包括与参考序列的指定位置相对应的位置。在这样的子集中,可以确定哪些(如果有的话)测序的核酸在指定位置处包含核苷酸变异,以及任选地哪些(如果有的话)包含参考核苷酸(即,与参考序列中的相同)。如果包含核苷酸变异的子集中测序的核酸的数目超过阈值,则变异核苷酸可以在指定位置处被识别。阈值可以是简单的数字,诸如包含核苷酸变异的子集中的至少1个、2个、3个、4个、5个、6个、7个、8个、9个或10个测序的核酸,或者阈值可以是包含核苷酸变异的子集中测序的核酸的比率,诸如至少0.5%、1%、2%、3%、4%、5%、10%、15%或20%以及其他可能性。可以对参考序列中感兴趣的任何指定位置重复比较。有时可以对占据参考序列上至少约20个、100个、200个或300个连续位置(例如,约20-500个或约50-300个连续位置)的指定位置进行比较。
[0100]
图1示出了用于检测和抑制比对错误的方法的实施方案。一般来说,该方法可以使用变异识别器(variant caller)和/或融合识别器来根据一组预定的指定阈值鉴定一组潜在的遗传变异。例如,变异识别器可用于根据指定阈值鉴定一组体细胞snv或插入或缺失变异,而融合识别器可用于根据指定阈值鉴定一组种系和体细胞基因内(基因之内)基因融合。这样一组潜在的遗传变异可以包括一个或更多个比对错误,其中当变异来源于加工的假基因的存在时,变异可能被错误地分配给基因(从而导致检测到假阳性遗传变异)。这样的比对错误可以诸如例如通过从对作为潜在遗传变异的鉴定或进一步分析中过滤或去除这种检测到的比对错误,在变异识别过程期间被检测和抑制。
[0101]
为了进一步说明本文公开的方法的方面,图2和图3提供了示意性描绘用于至少部分地使用计算机来产生过滤的序列信息数据集的示例性方法步骤的流程图。本文公开的任何方法可选地至少部分地在系统或计算机可读介质中实现或体现,这也在本文中进一步描述。如图2和图3中示出的,方法200和方法300都包括鉴定从获自受试者的生物样品中的cfdna分子或片段获得的测试序列读段的集合中的分裂序列读段,其中每个分裂序列读段在步骤202和步骤302中分别包含至少一个断点。典型地,方法200和方法300各自包括在步骤202和步骤302之前接收(例如,经由电子通信网络或其他通信或存储介质)测试序列信息,测试序列信息包括来自从受试者获得的生物样品中的cfdna分子的测试序列读段。在一些实施方案中,方法200和方法300各自包括在步骤202和步骤302之前对从受试者获得的生物样品中的cfdna片段进行测序,以产生测试序列读段的集合(即,测试序列信息)。
[0102]
任选地,使用多种技术中的任何一种或更多种,鉴定从样品获得的测试序列信息中的分裂序列读段或比对错误。在一些实施方案中,通过鉴定一组测试序列信息中的测试序列读段来鉴定分裂序列读段,该测试序列信息仅与一组给定参考序列信息部分地比对。
例如,分裂序列读段通常包括至少第一子序列和至少第二子序列,第一子序列映射到给定参考基因组序列的第一区域,第二子序列映射到给定参考基因组序列的第二区域,其中给定参考基因组序列的第一区域和第二区域彼此不连续或不相邻。在这些实施方案中的一些中,方法包括鉴定与第一断点相邻的第一子序列,第一断点映射到第一基因位点(例如,给定参考基因组序列的基因内基因座或基因间基因座),以及鉴定与第二断点相邻的第二子序列,第二断点映射到第二不同的基因位点(例如,给定参考基因组序列的非连续基因内基因座或非连续基因间基因座)。在这些实施方案中,第一断点和第二断点形成断点对。
[0103]
在其他示例性实施方案中,通过鉴定在测试序列信息中观察到的基因组区域(例如,编码序列(cds)等)相对于缺乏包含基因组区域的分裂序列读段的参考序列信息的增加的覆盖来鉴定给定分裂序列读段或比对错误。在一些实施方案中,怀疑的分裂序列或基因融合(例如,加工的假基因(ppg))不被如此识别,除非至少两个(例如2个、3个、4个、5个、6个、7个、8个、9个、10个或更多个)分裂序列读段各自包含至少一个相同的断点,但是在给定特性(诸如在长度方面)上彼此不同,这可以指示分裂序列读段来源于给定样品中不同的cfdna片段。这通常增加了在给定样品中观察到真正的分裂序列或基因融合的置信水平。可选地修改以用于本公开内容的方法和相关方面的关于鉴定分裂序列读段和基因融合的另外细节在例如wo 2017/062970和wo 2018/213814中提供,将其各自通过引用并入。
[0104]
同样如图2和图3中所示,方法200包括:在步骤204中,在测试序列读段的集合中,抑制包含距离给定断点选定数目的核苷酸内的至少一个序列变异的一个或更多个分裂序列读段的至少一部分(例如,给定读段的至少一部分和/或全部读段的至少一部分)和/或一个或更多个测试序列读段的至少一部分(例如,给定读段的至少一部分和/或全部读段的至少一部分),以产生过滤的序列信息数据集;而方法300包括:在步骤304中,在测试序列读段的集合中,抑制包含距离给定断点选定数目的核苷酸内的至少一个序列变异的分裂序列读段的一个或更多个碱基识别和/或测试序列读段的一个或更多个碱基识别,以产生过滤的序列信息数据集。序列读段(或其部分)和/或碱基识别通常通过从给定数据集去除该信息或仅通过在数据集的给定应用中不使用该信息来进行“抑制”。在一些示例性实施方案中,如本文描述的,被抑制的分裂序列读段包含加工的假基因(ppg)的至少一部分。
[0105]
在一些实施方案中,给定断点的选定数目的核苷酸内的序列变异包括的突变等位基因分数(maf)小于或等于生物样品中的断点的maf。尽管可选地使用其他数目,但是距离给定断点选定数目的核苷酸通常包括约1个、2个、3个、4个、5个、6个、7个、8个、9个、10个、11个、12个、13个、14个、15个、16个、17个、18个、19个、20个或更多个核苷酸。在其他实施方案中,距离给定断点的核苷酸数目可以包括少于100个、50个、20个、15个、10个、8个、6个、4个或2个核苷酸。另外,距离给定断点选定数目的核苷酸位于给定断点的5’和/或3’(即,在给定断点的任一侧或两侧)。如本文描述的,各种类型的序列变异可选地用于实施本公开内容的方法。在这些实施方案中的一些中,例如,序列变异包括单核苷酸变异(snv)和/或插入或缺失(indel)。在某些实施方案中,方法包括当另外的测试序列读段与选自由以下组成的组的一个或更多个基因序列的至少一部分对齐时,抑制包含不在距离给定断点选定数目的核苷酸内的一个或更多个序列变异的一个或更多个另外的测试序列读段或其部分:smad4、gnas、tp53、raf1、cdk4、tyro3、mapk1、stk11、ccnd1、hras、met、myc和nras。
[0106]
使用本文公开的方法产生的过滤的序列信息数据集可以用于许多应用。通常,它
们用于促进鉴定从受试者获得的测试样品中具有临床意义的序列变异,以确定受试者是否患有特定的疾病、紊乱或状况。在某些实施方案中,在特定的疾病、紊乱或状况被如此诊断后,方法还包括向受试者施用一种或更多种疗法以治疗受试者的该疾病、紊乱或状况,如本文进一步描述的。
[0107]
可以通过使用液体活组织检查测定来鉴定无细胞dna(例如,其包括循环肿瘤dna,即ctdna)中的体细胞基因组改变,从而从受试者的样品中鉴定基因融合。这样的测定可以包括对无细胞dna分子测序以产生序列读段,并使用基因标志物(例如,alk、fgfr2、fgfr3、ntrk1、ret和ros1)的组来分析序列读段。
[0108]
ppg在起源上可以是种系或体细胞的,并且可以通过分析遍及基因组的一个或更多个遗传位点处的序列读段覆盖数据来鉴定。例如,ppg可以发现于在跨外显子-外显子接点观察到比对假象的位置。ppg的存在可以通过缺少内含子序列的多个软剪读段(即,序列读段的一部分不与参考序列对齐的那些读段)的存在来揭示,或者通过内含子-外显子边界处覆盖的不连续性来揭示。ppg可以源自例如smad4、gnas、tp53、raf1、cdk4、tyro3、mapk1、stk11、ccnd1、hras、met、myc和nras的外显子序列。
[0109]
可以使用一个或更多个标准来鉴定潜在的比对错误。例如,在对应于包含基因内融合断点的基因融合的序列读段(基因融合读段)的集合中,可以从重叠覆盖基因融合的读段(包含在包含基因内融合断点的区域内的遗传变异)的子集中鉴定潜在的比对错误。该区域可以包含与基因内融合断点相邻的20个或更少的核苷酸(例如,约20个、15个、10个、8个、6个、4个或2个核苷酸)。基因融合读段的集合可以对应于一个或更多个加工的假基因(ppg),诸如样品特异性ppg(其对于特定的样品或受试者是特异性的,并且通常不存在于参考人类基因组诸如hg19中)。遗传变异可以包括单核苷酸变异(snv)或者插入或缺失(indel)。例如,snv可以位于内含子-外显子边界处或位于基因编码序列(cds)内。
[0110]
作为另一实例,在对应于基因融合的序列读段(基因融合读段)的集合中,可以从在smad4、tyro3和/或raf1基因中检测到的基因融合读段的子集中鉴定潜在的比对错误。
[0111]
当检测真正的遗传变异(例如,来自受试者的样品的无细胞dna分子)时,可以抑制已经鉴定出的潜在比对错误。例如,可以从基因融合读段的集合中过滤掉这样鉴定出的潜在比对错误的至少一部分,以产生过滤的序列读段。然后,可以处理或分析这样过滤的序列读段,以检测与参考序列相比的真正的遗传变异(例如,不是由于ppg的存在而导致的假阳性变异引起的),从而有利地降低变异的假阳性检测率。因此,可以通过以更高的准确度、灵敏度、特异性、阳性预测值(ppv)、阴性预测值(npv)或曲线下面积(auc)分析来自受试者的样品来鉴定变异。
[0112]
在一些情况下,检测到的比对错误的一部分基于在样品中检测到的比对错误具有的突变等位基因分数(maf)小于或等于对应于样品中的基因内融合断点的基因内融合的突变等位基因分数被过滤掉。因为融合介导的错误可以在跨融合读段中被发现,所以样品中的假阳性比对错误具有的maf不可能大于对应于样品中的基因内融合断点的基因内融合的maf。
[0113]
在一些情况下,检测到的比对错误的一部分基于基因融合读段包含不属于临床可操作变异的预定义集合的遗传变异被过滤掉。这样的“白名单”变异可见于多种变异数据库中,这些变异在受试者样品中的存在已被示出与受试者的疾病或紊乱(例如,癌症)相关或
指示该疾病或紊乱。这样的变异数据库可以包括,例如,癌症体细胞突变目录(catalogue of somatic mutations in cancer,cosmic)、癌症基因组图谱(tcga)和外显子组聚合联盟(exac)。由于它们与临床决策(例如,诊断、预后、治疗选择、靶向治疗、治疗监测、复发监测等)的相关性,这样的编目的变异的预定义集合可被指定用于进一步的生物信息学分析。这样的预定义集合可以基于例如对临床样品(例如,已知存在或不存在疾病或紊乱的患者组群的临床样品)的分析以及来自公共数据库和临床文献的注释信息来确定。
[0114]
在鉴定和抑制比对错误后,可以对过滤的序列读段的集合进行分析,以检测与参考序列相比的真正的遗传变异。
[0115]
本公开内容还提供了本文公开的方法步骤可选地被修改以用于使用本文公开的系统和/或计算机可读介质来执行。在某些方面,系统可以包括控制器,该控制器包括或能够访问包括非暂时性计算机可执行指令的计算机可读介质,当该指令被至少一个电子处理器执行时执行本文描述的至少一种方法。
[0116]
在一些实施方案中,测序仪是dna测序仪。在一些实施方案中,测序仪被设计成执行高通量测序,诸如下一代测序。在一些实施方案中,系统在测序仪中包括衔接子标记的cfdna分子。在一些实施方案中,衔接子标记的cfdna分子来源于一个受试者或多于一个受试者。在一些实施方案中,来自样品的cfdna分子带有独特的条形码或非独特的条形码。
[0117]
在一些实施方案中,由计算机处理器实施的方法还包括将序列读段分组为家族,每个家族包含含有相同条形码并具有相同的起始位置和终止位置的序列读段,由此每个家族包含从相同原始cfdna分子扩增的序列读段。
[0118]
在一些实施方案中,本文描述的方法和系统利用数字处理设备。在另外的实施方案中,数字处理设备包括执行设备功能的一个或更多个硬件中央处理单元(cpu)或通用图形处理单元(gpgpu)。在又一些另外的实施方案中,数字处理设备还包括被配置为执行可执行指令的操作系统。在一些实施方案中,数字处理设备可选地连接至计算机网络。在另外的实施方案中,数字处理设备可选地连接至互联网,使得它访问万维网。在又一些另外的实施方案中,数字处理设备可选地连接至云计算基础设施。在其他实施方案中,数字处理设备可选地连接至内联网。在其他实施方案中,数字处理设备可选地连接至数据存储设备。根据本文的描述,通过非限制性实例的方式,合适的数字处理设备包括服务器计算机、台式计算机、膝上型计算机、笔记本计算机、手持计算机、互联网设备、移动智能电话和平板计算机。
[0119]
在一些实施方案中,数字处理设备包括被配置为执行可执行指令的操作系统。例如,操作系统是软件,包括程序和数据,其管理设备的硬件并提供用于执行应用的服务。本领域技术人员将认识到,通过非限制性实例的方式,合适的服务器操作系统包括freebsd、openbsd、linux、mac os xwindows和和本领域技术人员将认识到,通过非限制性实例的方式,合适的个人计算机操作系统包括mac osos和unix型操作系统,例如在一些实施方案中,操作系统由云计算提供。本领域技术人员还将认识到,通过非限制性实例的方式,合适的移动智能电话操作系统包括os、os、research inblackberry
windowsos、windowsos、和
[0120]
在一些实施方案中,该设备包括存储和/或存储器设备。存储和/或存储器设备是用于在临时或永久基础上存储数据或程序的一个或更多个物理装置。在一些实施方案中,该设备是易失性存储器并且需要电力来维持所存储的信息。在一些实施方案中,该设备是非易失性存储器并且当数字处理设备未被供电时保留所存储的信息。在另外的实施方案中,非易失性存储器包括闪存。在一些实施方案中,非易失性存储器包括动态随机存取存储器(dram)。在一些实施方案中,非易失性存储器包括铁电随机存取存储器(fram)。在一些实施方案中,非易失性存储器包括相变随机存取存储器(pram)。在其他实施方案中,该设备是存储设备,通过非限制性实例的方式包括cd-rom、dvd、闪存设备,磁盘驱动器、磁带驱动器、光盘驱动器以及基于云计算的存储器。在另外的实施方案中,存储和/或存储器设备是诸如本文公开的那些设备的组合的设备。
[0121]
在一些实施方案中,数字处理设备包括将视觉信息发送给用户的电子显示器。在一些实施方案中,显示器是液晶显示器(lcd)。在另外的实施方案中,显示器是薄膜晶体管液晶显示器(tft-lcd)。在一些实施方案中,显示器是有机发光二极管(oled)显示器。在各种另外的实施方案中,在oled显示器上是无源矩阵oled(pmoled)或有源矩阵oled(amoled)显示器。在一些实施方案中,显示器是等离子体显示器。在其他实施方案中,显示器是视频投影机。在另外其他实施方案中,显示器是与数字处理设备通信的头戴式显示器,诸如vr耳机。在另外的实施方案中,通过非限制性实例的方式,合适的vr耳机包括htc vive、oculus rift、samsung gear vr、microsoft hololens、razer osvr、fove vr、zeiss vr one、avegant glyph、freefly vr耳机等。在又一些另外的实施方案中,显示器是诸如本文公开的那些设备的组合的设备。
[0122]
在一些实施方案中,数字处理设备包括用于从用户接收信息的输入设备。在一些实施方案中,输入设备是键盘。在一些实施方案中,输入设备是定点设备,通过非限制性实例的方式包括鼠标、轨迹球、触控板、操纵杆、游戏控制器或指示笔。在一些实施方案中,输入设备是触摸屏或多点触摸屏。在其他实施方案中,输入设备是用于捕获语音或其他声音输入的麦克风。在其他实施方案中,输入设备是捕获运动或视觉输入的摄像机或其他传感器。在另外的实施方案中,输入设备是kinect、leap motion等。在又一些另外的实施方案中,输入设备是诸如本文公开的那些设备的组合的设备。
[0123]
在一些方面,本公开内容提供了一种系统,该系统包括控制器,该控制器包括或能够访问包括非暂时性计算机可执行指令的计算机可读介质,当该指令被至少一个电子处理器执行时执行本文提供的方法。
[0124]
图5示出了被编程或以其他方式配置以实现本文提供的方法的计算机系统501。
[0125]
计算机系统501可以被编程或以其他方式配置成实现用于检测和/或抑制基因序列读段中的比对错误的方法。计算机系统501可以调节本公开内容的多个方面,例如,(a)对生物样品中的无细胞核酸分子进行测序以产生基因序列读段;(b)将基因序列读段与参考序列比对以产生比对的序列读段;(c)从比对的序列读段中鉴定包含基因内融合断点的基因融合读段的集合;(d)通过鉴定在包含基因内融合断点的区域内包含遗传变异的基因融合读段的一个或更多个的子集来检测比对错误,其中该区域包含与基因内融合断点相邻的
一个或更多个核苷酸;(e)过滤掉基因融合读段的子集中的检测到的比对错误的至少一部分,以产生过滤的序列读段;以及(f)检测与参考序列相比包含真正遗传变异的过滤的序列读段。计算机系统501可以是用户的电子设备或相对于该电子设备远程定位的计算机系统。电子设备可以是移动电子设备。
[0126]
计算机系统501包括中央处理单元(cpu,在本文中也被称为“处理器”和“计算机处理器”)505,中央处理单元505可以是单核或多核处理器或用于并行处理的多于一个处理器。计算机系统501还包括存储器或存储器位置510(例如,随机存取存储器、只读存储器、闪速存储器)、电子存储单元515(例如,硬盘)、用于与一个或更多个其他系统进行通信的通信接口520(例如,网络适配器)和外围设备525,诸如高速缓冲存储器、其他存储器、数据存储和/或电子显示适配器。存储器510、存储单元515、接口520和外围设备525与cpu 505通过通信总线(实线)诸如主板(motherboard)通信。存储单元515可以是用于存储数据的数据存储单元(或数据储存库)。计算机系统501可以借助于通信接口520被可操作地耦合至计算机网络(“网络”)530。网络530可以是因特网(internet)、互联网(internet)和/或外联网、或与因特网通信的内联网和/或外联网。在一些情况下,网络530为电信和/或数据网络。网络530可以包括一个或更多个计算机服务器,这可以支持分布式计算,诸如云计算。在一些情况下,借助于计算机系统501,网络530可以实现对等网络(peer-to-peer network),其可以使耦合至计算机系统501的设备能够作为客户端或服务器运行。
[0127]
cpu 505可以执行一系列的机器可读指令,该机器可读指令可以以程序或软件来体现。指令可以被存储于存储器位置诸如存储器510中。指令可以被导向cpu 505,其可以随后编程或以其他方式配置cpu 505,以实现本公开内容的方法。cpu 505进行的操作的实例可以包括读取、解码、执行和写回。
[0128]
cpu 505可以是电路诸如集成电路的一部分。系统501的一个或更多个其他组件可以被包括在该电路中。在一些情况下,电路为专用集成电路(asic)。
[0129]
存储单元515可以存储文件,诸如驱动、库和保存的程序。存储单元515可以存储用户数据,例如,用户偏好和用户程序。在一些情况下,计算机系统501可以包括在计算机系统501的外部的一个或更多个另外的数据存储单元,诸如位于通过内联网或因特网而与计算机系统501通信的远程服务器上。
[0130]
计算机系统501可以与一个或更多个远程计算机系统通过网络530进行通信。例如,计算机系统501可以与用户的远程计算机系统进行通信。远程计算机系统的实例包括个人计算机(例如便携式pc)、板型或平板pc(例如ipad、galaxy tab)、电话、智能电话(例如iphone、android支持的设备、)或个人数字助理。用户可以经由网络530访问计算机系统501。
[0131]
如本文描述的方法可以通过机器(例如,计算机处理器)可执行代码的方式实现,该机器可执行代码被存储在计算机系统501的电子存储位置,诸如例如存储器510或电子存储单元515上。机器可执行代码或机器可读代码可以以软件的形式提供。在使用期间,代码可以由处理器505执行。在一些情况中,代码可以从存储单元515检索并存储在存储器510上,用于由处理器505迅速访问。在一些情况中,可以排除电子存储单元515,而机器可执行指令被存储于存储器510中。
[0132]
代码可以被预编译并配置为用于与具有适用于执行该代码的处理器的机器一起使用,或者可以在运行时间期间被编译。代码可以以编程语言的形式提供,该编程语言可被选择以使代码能够以预编译的或按编译原样(as-compiled)的方式被执行。
[0133]
本文所提供的系统和方法的方面,诸如计算机系统501,可以以编程来体现。技术的多个方面可以被认为是通常呈一种机器可读介质进行或体现的机器(或处理器)可执行代码和/或相关数据的形式的“产品”或“制品(articles of manufacture)”。机器可执行代码可以被存储于电子存储单元诸如存储器(例如,只读存储器、随机存取存储器、闪速存储器)或硬盘上。“存储”型介质可以包括计算机、处理器等的任何或所有有形存储器,或其相关模块,诸如多种半导体存储器、磁带驱动器、磁盘驱动器等,其可以在任何时间为软件编程提供非暂时性存储。软件的所有或部分有时可以通过因特网或多种其他电信网络进行通信。例如,此类通信可以使得将软件从一个计算机或处理器加载到另一个计算机或处理器,例如,从管理服务器或主机加载到应用服务器的计算机平台。因此,能够携带软件元件的另一类型的介质包括诸如在本地设备之间的物理接口、通过有线和光纤陆线网络以及在多种空中链路(air-links)上使用的光波、电波和电磁波。携带此类波的物理元件,诸如有线或无线链路、光链路等,也可以被认为是携带软件的介质。如本文使用的,除非被限制为非暂时性的、有形的“存储”介质,否则术语诸如计算机或机器“可读介质”指参与将指令提供至处理器用于执行的任何介质。
[0134]
因此,机器可读介质,诸如计算机可执行代码,可以采取多种形式,包括但不限于有形存储介质、载波介质或物理传输介质。非易失性存储介质包括,例如光盘或磁盘,诸如在任何计算机等中的任何存储设备,诸如可用于实现如附图中示出的数据库等。易失性存储介质包括动态存储器,诸如此类计算机平台的主存储器。有形的传输介质包括同轴电缆;铜线和光纤,包括构成计算机系统内的总线的导线。载波传输介质可以采取电信号或电磁信号或者声波或光波的形式,诸如在射频(rf)和红外(ir)数据通信期间产生的那些。因此,计算机可读介质的常见形式包括,例如:软盘(floppy disk)、软性磁盘(flexible disk)、硬盘、磁带、任何其他磁介质、cd-rom、dvd或dvd-rom、任何其他光学介质、穿孔卡片纸带、具有打孔模式的任何其他物理存储介质、ram、rom、prom和eprom、flash-eprom、任何其他存储器芯片或盒、传输数据或指令的载波、传输此类载波的缆线或链路,或者计算机可以从其读取编程代码和/或数据的任何其他介质。计算机可读介质的这些形式中的许多形式可以参与向处理器传送一个或更多个指令的一个或更多个序列以用于执行。
[0135]
计算机系统501可以包括电子显示器535或者与电子显示器535通信,电子显示器535包括用户界面(ui)540。ui的实例包括但不限于图形用户界面(gui)和基于网络的用户界面。
[0136]
本公开内容的方法和系统可以通过一个或更多个算法来实现。算法可以在由中央处理单元505执行后通过软件来实现。该算法可以,例如,(a)对生物样品中的无细胞核酸分子进行测序以产生基因序列读段;(b)将基因序列读段与参考序列比对以产生比对的序列读段;(c)从比对的序列读段中鉴定包含基因内融合断点的基因融合读段的集合;(d)通过鉴定在包含基因内融合断点的区域内包含遗传变异的基因融合读段的一个或更多个的子集来检测比对错误,其中该区域包含与基因内融合断点相邻的一个或更多个核苷酸;(e)过滤掉基因融合读段的子集中的检测到的比对错误的至少一部分,以产生过滤的序列读段;
以及(f)检测与参考序列相比包含真正遗传变异的过滤的序列读段。
[0137]
ii.方法的一般特征
[0138]
a.样品
[0139]
样品可以是从受试者分离的任何生物样品。样品可以包括身体组织,诸如已知或怀疑的实体瘤、全血、血小板、血清、血浆、粪便、红细胞、白细胞(white blood cell)或白细胞(leucocyte)、内皮细胞、组织活组织检查、脑脊液、滑液、淋巴液、腹水、组织液或细胞外液、细胞间的空间中的液体包括龈沟液、骨髓、胸腔积液、脑脊液(csf)、唾液、粘液、痰、精液、汗液、尿液。样品优选为体液,特别是血液及其级分,以及尿液。样品还可以包括从肿瘤脱落的核酸,例如循环肿瘤dna(ctdna)。核酸可以包括dna和rna,并且可以是双链和单链形式。样品可以是最初从受试者分离的形式,或者可以经过进一步处理以:去除或添加组分,诸如细胞;相对于一种组分富集另一种组分;或者将一种形式的核酸转化为另一种形式,诸如将rna转化为dna或将单链核酸转化为双链核酸。因此,例如,用于分析的体液是含有无细胞核酸的血浆或血清,例如,无细胞dna(cfdna)。
[0140]
血浆的体积可以取决于测序区域所期望的读段深度。示例性体积为0.4-40ml、5-20ml和10-20ml。例如,体积可以是0.5ml、1ml、5ml、10ml、20ml、30ml或40ml。取样血浆的体积可以是5ml至20ml。
[0141]
样品可以包含不同量的包含基因组当量的核酸。例如,约30ng dna的样品可以包含约10,000(104)个单倍体人类基因组当量,而在cfdna的情况下,可以包含约2000亿(2x10
11
)个单独的多核苷酸分子。类似地,约100ng dna的样品可以包含约30,000个单倍体人类基因组当量,而在cfdna的情况下,可以包含约6000亿个单独的分子。
[0142]
样品可以包含来自不同来源的核酸,例如,来自无细胞核酸或来自外来物(foreign object)的核酸。样品可以包含携带突变的核酸。例如,样品可以包含携带种系突变和/或体细胞突变的dna。样品可以包含携带癌症相关突变(例如,癌症相关的体细胞突变)的dna。
[0143]
在扩增前,样品中无细胞核酸的示例性量的范围为约1飞克(fg)至约1微克(ug),例如,1皮克(pg)至200纳克(ng)、1ng至100ng、10ng至1000ng。例如,量可以多达约600ng、多达约500ng、多达约400ng、多达约300ng、多达约200ng、多达约100ng、多达约50ng或多达约20ng的无细胞核酸分子。量可以是至少1fg、至少10fg、至少100fg、至少1pg、至少10pg、至少100pg、至少1ng、至少10ng、至少100ng、至少150ng或至少200ng的无细胞核酸分子。量可以多达1飞克(fg)、10fg、100fg、1皮克(pg)、10pg、100pg、1ng、10ng、100ng、150ng或200ng的无细胞核酸分子。方法可以包括获得1飞克(fg)至200fg。
[0144]
在某些实施方案中,样品中无细胞核酸的量在约5ng至300ng之间。
[0145]
无细胞核酸具有约100-500个核苷酸的示例性尺寸分布,其中110个至约230个核苷酸的分子代表约90%的分子,众数为约168个核苷酸,并且第二次峰值在240个至440个核苷酸范围内。无细胞核酸可以是约160个至约180个核苷酸,或约320个至约360个核苷酸,或约440个至约480个核苷酸。
[0146]
无细胞核酸可以通过分区步骤(partitioning step)从体液分离,在该分区步骤中,如在溶液中存在的无细胞dna被与细胞和体液的其他不可溶性组分分开。分区可以包括诸如离心或过滤的技术。可选地,体液中的细胞可以被裂解,并且无细胞核酸和细胞核酸可
以被一起处理。通常,在添加缓冲液和洗涤步骤之后,无细胞核酸可以用醇沉淀。可以使用进一步的清洁步骤诸如基于二氧化硅的柱来去除污染物或盐。例如,可以在整个反应中添加非特异性批量(bulk)载体核酸以优化该程序的某些方面诸如收率。在这样的处理之后,样品可以包括各种形式的核酸,包括双链dna、单链dna和单链rna。任选地,单链dna和rna可以被转化成双链形式,使得它们被包括在随后的处理和分析步骤中。
[0147]
b.标签
[0148]
提供样品索引和/或分子条形码的标签可以通过化学合成、连接、重叠延伸pcr和其他方法掺入衔接子中或另外连接至衔接子。通常,反应中独特或非独特的分子条形码的布置遵循美国专利申请20010053519、20110160078和美国专利第6,582,908号、第7,537,898号和第9,598,731号描述的方法和系统。
[0149]
标签可以随机或非随机地连接至样品核酸。在一些情况下,它们以预期比率被引入。条形码的集合可以是独特的,例如,所有的条形码具有相同的核苷酸序列。条形码的集合可以是非独特的,例如,条形码中的一些具有相同的核苷酸序列,而条形码中的一些具有不同的核苷酸序列。例如,可以加载标识符使得每个基因组样品加载多于1个、2个、3个、4个、5个、6个、7个、8个、9个、10个、20个、50个、100个、500个、1000个、5000个、10000个、50,000个、100,000个、500,000个、1,000,000个、10,000,000个、50,000,000个或1,000,000,000个标识符。在一些情况下,可以加载标识符使得每个基因组样品加载少于2个、3个、4个、5个、6个、7个、8个、9个、10个、20个、50个、100个、500个、1000个、5000个、10000个、50,000个、100,000个、500,000个、1,000,000个、10,000,000个、50,000,000个或1,000,000,000个标识符。在一些情况下,每个样品基因组加载的标识符的平均数目少于或大于每个基因组样品约1个、2个、3个、4个、5个、6个、7个、8个、9个、10个、20个、50个、100个、500个、1000个、5000个、10000个、50,000个、100,000个、500,000个、1,000,000个、10,000,000个、50,000,000个或1,000,000,000个标识符。
[0150]
一种优选的形式使用20-50个不同的标签,连接至靶分子的两端,产生20-50
×
20-50个标签。标签的这种数目足以使具有相同的起始点和终止点的不同分子具有高概率(例如,至少94%,99.5%,99.99%,99.999%)接收不同的标签组合。
[0151]
在一些情况下,标识符可以是预定的或随机的或半随机的序列的寡核苷酸。在其他情况下,可以使用多于一个条形码,使得条形码在所述多于一个条形码中相对于彼此不必是独特的。在该实例中,条形码可以附接(例如,通过连接或pcr扩增)至单独的分子,使得条形码与其可以附接的序列的组合产生可以被单独追溯的独特的序列。如本文描述的,检测非独特的条形码与映射到参考序列或基因组的序列读段的开始(起始)和结束(终止)部分的序列数据的组合可以允许将独特的身份分配至特定分子。单独序列读段的长度或碱基对数目也可以用于将独特身份分配至此类分子。如本文描述的,来自已经被分配了独特身份的核酸的单链的片段可以从而允许随后鉴定来自该亲本链和/或互补链的片段。
[0152]
使用常规的核酸扩增方法,可以应用一个或更多个扩增以便将分子条形码和/或样品索引引入核酸分子中。扩增可以在一种或更多种反应混合物中进行。分子条形码和样品索引可以同时引入或以任何顺序引入。分子条形码和样品索引可以在序列捕获(例如富集)之前和/或之后引入。在一些实施方案中,在探针捕获之前仅引入分子标签,而在序列捕获之后引入样品索引/标签。在一些情况下,分子条形码和样品索引都是在探针捕获之前引
入的。在一些情况下,样品索引在序列捕获之后引入。通常,序列捕获包括引入与靶向序列互补的单链核酸分子。通常,扩增产生多于一个非独特或独特地加标签的核酸扩增子,其分子条形码和样品索引的尺寸范围为200个核苷酸(nt)至700nt、250nt至350nt或320nt至550nt。在一些实施方案中,扩增子具有约300nt的尺寸。在一些实施方案中,扩增子具有约500nt的尺寸。
[0153]
c.扩增
[0154]
侧翼有衔接子的样品核酸可以通过pcr和其他扩增方法进行扩增,这些方法通常从与待扩增核酸分子侧翼的衔接子中的引物结合位点结合的引物引发。扩增方法可以包括由热循环引起的延伸、变性和退火的循环,或者可以是等温的,如在转录介导的扩增中。其他扩增方法包括连接酶链式反应、链置换扩增、基于核酸序列的扩增和基于自身持续序列的复制。
[0155]
d.富集
[0156]
测序前可以富集序列。可以对特定靶区域进行富集或对(“靶序列”)进行非特异性地富集。在一些实施方案中,感兴趣的靶向区域可以用针对一个或更多个诱饵集组选择的捕获探针(“诱饵”)使用差异性平铺和捕获方案来富集。差异性平铺和捕获方案使用不同相对浓度的诱饵集在与诱饵相关的基因组区域中差异性平铺(例如,以不同的“分辨率”),经受一组限制(例如,测序仪限制,诸如测序载量、每种诱饵的效用等),并以下游测序所需的水平捕获它们。在一些实施方案中,具有针对一个或更多个感兴趣区域的探针的生物素标记的珠可以用于捕获靶序列,任选地随后扩增这些区域,以富集感兴趣区域。
[0157]
序列捕获通常包括使用与靶序列杂交的寡核苷酸探针。探针集策略可以包括将探针平铺在感兴趣的区域内。这样的探针可以是,例如,长度约60个至120个碱基。探针集可以具有约2x、3x、4x、5x、6x、8x、9x、10x、15x、20x、50x或更大的深度。序列捕获的有效性部分地取决于靶分子中与探针序列互补(或几乎互补)的序列的长度。
[0158]
e.测序
[0159]
可以对侧翼有衔接子、有或没有预先扩增的样品核酸进行测序。测序方法包括:例如,sanger测序、高通量测序、焦磷酸测序、合成测序、单分子测序、纳米孔测序、半导体测序、连接测序、杂交测序、rna-seq(illumina)、数字基因表达(helicos)、下一代测序、单分子合成测序(smss)(helicos)、大规模并行测序、克隆单分子阵列(solexa)、鸟枪法测序、ion torrent、牛津纳米孔(oxford nanopore)、roche genia、maxim-gilbert测序、引物步移、使用pacbio、solid、ion torrent或纳米孔平台的测序。测序反应可以在多种样品处理单元中进行,这些样品处理单元可以是多行道(multiple lane)、多通道、多孔或基本上同时处理多个样品集合的其他装置。样品处理单元还可以包括多个样品室,以便能够同时处理多个运行。
[0160]
可以对一种或更多种已知包含癌症或其他疾病的标志物的片段类型进行测序反应。也可以对样品中存在的任何核酸片段进行测序反应。测序反应可以提供基因组的至少5%、10%、15%、20%、25%、30%、40%、50%、60%、70%、80%、90%、95%、99%、99.9%或100%的序列覆盖度。在其他情况下,基因组的序列覆盖度可以小于5%、10%、15%、20%、25%、30%、40%、50%、60%、70%、80%、90%、95%、99%、99.9%或100%。
[0161]
可以使用多重测序进行同时测序反应。在一些情况下,可以用至少1000个、2000
个、3000个、4000个、5000个、6000个、7000个、8000个、9000个、10000个、50000个或100,000个测序反应对无细胞多核苷酸进行测序。在其他情况下,可以用少于1000个、2000个、3000个、4000个、5000个、6000个、7000个、8000个、9000个、10000个、50000个或100,000个测序反应对无细胞多核苷酸进行测序。测序反应可以顺序进行或同时进行。可以对所有或部分的测序反应进行后续数据分析。在一些情况下,可以对至少1000个、2000个、3000个、4000个、5000个、6000个、7000个、8000个、9000个、10000个、50000个或100,000个测序反应进行数据分析。在其他情况下,可以对少于1000个、2000个、3000个、4000个、5000个、6000个、7000个、8000个、9000个、10000个、50000个或100,000个测序反应进行数据分析。示例性的读段深度是每个基因座(碱基)1000-50000个读段。
[0162]
f.分析
[0163]
根据本公开内容的实施方案的测序产生多于一个序列读段。根据本公开内容的序列读段通常包括长度小于约150个碱基,或长度小于约90个碱基的核苷酸序列数据。在某些实施方案中,读段长度为约80个和约90个碱基之间,例如约85个碱基。在一些实施方案中,本公开内容的方法适用于非常短的读段,即长度小于约50个或约30个碱基。序列读段数据可以包括序列数据以及元信息。如本领域技术人员已知的,序列读段数据可以以任何合适的文件格式存储,包括例如vcf文件、fasta文件或fastq文件。
[0164]
fasta最初是一个用于检索序列数据库的计算机程序,并且名称fasta变为也指标准文件格式。参见pearson&lipman,1988,improved tools for biological sequence comparison,pnas 85:2444-2448。fasta格式的序列以单行描述开始,随后为序列数据行。描述行通过第一列中的大于(“>”)符号与序列数据区分开。“>”符号后面的词是序列的标识符,并且该行的其余部分是描述(两者都是任选的)。在“>”和标识符的第一个字母之间不应当有空格。建议所有文本行都短于80个字符。如果出现以“>”开头的另一行,则序列结束;这指示另一序列的开始。
[0165]
fastq格式是一种基于文本的格式,用于存储生物序列(通常是核苷酸序列)及其对应的质量评分。它与fasta格式相似,但是在序列数据之后具有质量评分。为了简洁,序列字母和质量评分都用单个ascii字符编码。fastq格式是用于存储高通量测序仪器诸如illumina genome analyzer的输出结果的约定俗成的标准。cock等人,2009,the sanger fastq file format for sequences with quality scores,and the solexa/illumina fastq variants,nucleic acids res 38(6):1767-1771。
[0166]
对于fasta和fastq文件,元信息包括描述行但不包括序列数据行。在一些实施方案中,对于fastq文件,元信息包括质量评分。对于fasta和fastq文件,序列数据在描述行之后开始,并且通常使用一些任选地带有
“-”
的iupac模糊代码的子集呈现。在优选实施方案中,序列数据将使用a、t、c、g和n字符,任选地根据需要包括
“-”
或“u”(例如,以表示空位或尿嘧啶)。
[0167]
在一些实施方案中,至少一个主序列读段文件和输出文件被存储为纯文本文件(例如,使用诸如ascii、iso/iec 646、ebcdic、utf-8或utf-16的编码)。本发明提供的计算机系统可以包括能够打开纯文本文件的文本编辑器程序。文本编辑器程序可以指能够在计算机屏幕上呈现文本文件(诸如纯文本文件)的内容、允许人员编辑文本(例如使用显示器、键盘和鼠标)的计算机程序。示例性的文本编辑器包括但不限于microsoft word、emacs、
pico、vi、bbedit和texgwrangler。优选地,文本编辑器程序能够以人类可读格式在计算机屏幕上显示纯文本文件,显示元信息和序列读段(例如,不是二进制编码而是使用字母数字字符,因为它们将在印刷人类书写中使用)。
[0168]
虽然已经参照fasta或fastq文件讨论了方法,但是本公开内容的方法和系统可以用于压缩任何合适的序列文件格式,包括例如变异识别格式(variant call format,vcf)格式的文件。一个典型的vcf文件将包括一个标题段和一个数据段。标题包含任何数目的元信息行(每行都以字符
‘
##’开始)和以单个
‘
#’字符开始的tab分隔字段定义行。字段定义行命名八个必填列,而正文段包含填充由字段定义行定义的列的数据行。vcf格式在danecek,2011,the variant call format and vcftools,bioinformatics 27(15):2156-2158中描述。标题段可被视为要写入压缩文件的元信息,而数据段可被视为行,其中每一行只有在为独特的情况下才会被存储在主文件中。
[0169]
本公开内容的某些实施方案提供了序列读段的装配。例如,在通过比对的装配中,读段彼此比对或与参考比对。通过比对每个读段,继而与参考基因组比对,所有读段被按照关于彼此的关系定位以创建装配体。另外,将序列读段与参考序列比对或映射到参考序列也可以用于鉴定序列读段中的变异序列。鉴定变异序列可以与本文描述的方法和系统组合使用,以进一步帮助疾病或状况的诊断或预后或用于指导治疗决策。
[0170]
在一些实施方案中,任何或所有步骤都是自动化的。可选地,本发明的方法可以全部或部分地体现在一个或更多个专用程序中,例如,每个程序可选地用诸如c++的编译语言编写,然后编译并以二进制形式分发。本发明的方法可以全部或部分地实现为现有序列分析平台内的模块,或者通过调用现有序列分析平台内的功能来实现。在某些实施方案中,本发明的方法包括多个步骤,这些步骤都响应于单个起始队列(例如,源自人类活动、另一计算机程序或机器的触发事件中的一个或组合)而被自动调用。因此,本发明提供了其中任何步骤或步骤的任何组合可以响应于队列而自动发生的方法。自动通常意味着没有干预人工输入、影响或交互(即,仅响应于原来或预先排队的人工活动)。
[0171]
该系统还包括多种形式的输出,所述多种形式的输出包括对受试者核酸的准确且灵敏的解释。检索的输出可以以计算机文件的格式提供。在某些实施方案中,输出是fasta文件、fastq文件或vcf文件。输出可以被处理以产生含有序列数据诸如与参考基因组的序列比对的核酸序列的文本文件或xml文件。在其他实施方案中,处理产生包含坐标或描述受试者核酸中相对于参考基因组的一个或更多个突变的字符串的输出。本领域已知的比对字符串包括simple ungapped alignment report(sugar)、verbose useful labeled gapped alignment report(valgar)和compact idiosyncratic gapped alignment report(cigar)(ning,z.等人,genome research 11(10):1725-9(2001))。这些字串例如在来自european bioinformatics institute(hinxton,uk)的exonerate序列比对软件中实现。
[0172]
在一些实施方案中,产生包含cigar字符串的序列比对——诸如,例如序列比对图(sam)或二元比对图(bam)文件(sam格式在例如li等人,the sequence alignment/map format and samtools,bioinformatics,2009,25(16):2078-9中描述)。在一些实施方案中,cigar显示或包括每行一个空位的比对。cigar是一种报告为cigar字符串的压缩的成对比对格式。cigar字符串可用于呈现长的(例如,基因组)成对比对。在sam格式中使用cigar字符串来表示读段与参考基因组序列的比对。
[0173]
cigar字符串可以遵循建立的基序。每个字符前面是数字,给出事件的碱基计数。使用的字符可以包括m、i、d、n和s(m=匹配;i=插入;d=缺失;n=空位;s=取代)。cigar字符串定义匹配/不匹配和缺失(或空位)的序列。例如,cigar字符串2md3m2d2m将意味着比对包含2个匹配、1个缺失(为了节省一些空间省略数字1)、3个匹配、2个缺失和2个匹配。
[0174]
如本发明所设想的,上文描述的功能可以使用包括软件、硬件、固件、硬连线或这些的任何组合的系统来实现。实现功能的特征也可以物理地位于不同位置处,包括被分布使得部分功能在不同物理位置处实现。
[0175]
系统可以包括服务器计算机、终端、测序仪、测序仪计算机、计算机或其任何组合中的一个或更多个。每个这样的计算机设备可以经由网络通信。测序仪可以任选地包括或可操作地耦合到其自身的例如专用的测序仪计算机(包括任何输入/输出机制(i/o)、处理器和存储器)。另外或可选地,测序仪可以经由网络可操作地耦合到服务器或计算机(例如,膝上型电脑、台式电脑或平板电脑)。计算机包括一个或更多个处理器、存储器和i/o。在本发明的方法采用客户端/服务器架构的情况下,本发明的方法的任何步骤可以使用服务器来执行,该服务器包括能够获得数据、指令等的处理器、存储器和i/o中的一个或更多个,或经由接口模块提供结果或以文件提供结果。服务器可以通过计算机或终端在网络上连接,或者服务器可以直接连接至终端。终端优选是计算机设备。根据本发明的计算机优选地包括耦合到i/o机制和存储器的一个或更多个处理器。
[0176]
处理器可以由一个或更多个处理器提供,处理器包括例如一个或更多个单核或多核处理器。i/o机制可以包括视频显示单元(例如,液晶显示器(lcd)或阴极射线管(crt))、字母数字输入设备(例如,键盘)、光标控制设备(例如,鼠标)、磁盘驱动单元、信号产生设备(例如,扬声器)、加速度计、麦克风、蜂窝射频天线和网络接口设备(例如,网络接口卡(nic)、无线网卡、蜂窝调制解调器、数据插孔、以太网端口、调制解调器插孔、hdmi端口、迷你hdmi端口、usb端口)、触摸屏(例如,crt、lcd、led、amoled、super amoled)、定点设备、触控板、灯(例如,led)、光/图像投影设备或其组合。根据本发明的存储器是指由一个或更多个有形设备提供的非暂时性存储器,该有形设备优选地包括一个或更多个机器可读介质,在该介质上存储一组或更多组指令(例如,软件),该指令体现了在本文中描述的任一种或更多种方法或功能。软件也可以在由系统内的计算机执行期间完全或至少部分地驻留在主存储器、处理器或两者中,主存储器和处理器也构成机器可读介质。软件还可以经由网络接口设备通过网络发送或接收。
[0177]
尽管机器可读介质在示例性实施方案中可以是单个介质,但是术语“机器可读介质”应该被认为包括存储一组或更多组指令的单个介质或多个介质(例如,集中式或分布式数据库和/或相关联的高速缓冲存储器和服务器)。术语“机器可读介质”还应该被认为包括能够存储、编码或携带指令集的任何介质,指令集用于由机器执行并使机器执行本发明的任一种或更多种方法。存储器可以是例如硬盘驱动器、固态驱动器(ssd)、光盘、闪存、压缩盘、磁带驱动器、“云”存储位置或其组合中的一个或更多个。在某些实施方案中,本发明的设备包括有形的、非暂时性的计算机可读存储介质。用作存储器的示例性设备包括半导体存储器设备(例如,eprom、eeprom、固态驱动器(ssd)以及闪存设备(例如,sd、micro sd、sdxc、sdio、sdhc卡));磁盘(例如,内部硬盘或可移动盘);和光盘(例如,cd盘和dvd盘)。
[0178]
在一些实施方案中,本文公开的系统和方法的结果被用作产生报告的输入。报告
可以是纸质形式的。例如,报告可以包括源自通过本文公开的方法和系统鉴定的过滤的序列信息的数据。这样的数据可以包括例如考虑所鉴定的序列信息的诊断信息或治疗建议。在一些实施方案中,报告可以包括信息,诸如通过本文公开的方法和系统鉴定的一个或更多个真正遗传变异。
[0179]
本文公开的方法的多种步骤或由本文公开的系统执行的步骤可以在相同或不同的时间、在相同或不同的地理位置(例如,国家)和/或由同一人或不同的人执行。
[0180]
iii.示例性应用
[0181]
a.测序组(sequencing panel)
[0182]
为了提高检测到指示肿瘤的突变的可能性,测序的dna区域可以包含一组基因或基因组区域。选择有限的测序区域(例如,有限的组)可以减少所需的总测序(例如,测序的核苷酸总量)。测序组可以靶向多于一个不同的基因或区域来检测单个癌症、癌症的集合或所有癌症。可选地,可以通过全基因组测序(wgs)或其他无偏倚的测序方法对dna进行测序,而不使用测序组。
[0183]
在一些方面,选择靶向多于一个不同基因或基因组区域的组,使得确定比例的患有癌症的受试者在该组中的一个或更多个不同基因中显示遗传变异或肿瘤标志物。可以选择组,以将测序区域限制在固定数目的碱基对。可以选择组,以对期望的量的dna测序。还可以选择组,以实现期望的序列读段深度。可以选择组,以针对一定量的测序碱基对实现期望的序列读段深度或序列读段覆盖度。可以选择组,以实现检测样品中的一种或更多种遗传变异的理论灵敏度、理论特异性和/或理论准确度。
[0184]
用于检测区域的组的探针可以包括用于检测感兴趣的基因组区域(热点区域)的探针以及核小体感知(nucleosome-aware)探针(例如,kras密码子12和13),并且可以被设计成基于对受核小体结合模式和gc序列组成影响的cfdna覆盖度和片段尺寸变化的分析来优化捕获。本文使用的区域还可以包括基于核小体位置和gc模型来优化的非热点区域。该组可以包括多于一个子组(subpanels),包括用于鉴定以下的子组:来源组织(例如,使用已公布的文献来定义50-100个诱饵,这些诱饵代表跨组织具有最多样的转录谱(不一定为启动子)的基因)、全基因组骨架(例如,用于鉴定超保守基因组含量,及为了拷贝数碱基排列(copy number base lining)的目的用少数探针跨染色体稀疏平铺)、转录起始位点(tss)/cpg岛(例如,用于捕获在例如肿瘤抑制基因(例如,结肠直肠癌中的sept9/vim)启动子中的差异甲基化区域(例如,差异性甲基化区域(dmr))。在一些实施方案中,来源组织的标志物是组织特异性的表观遗传标志物。
[0185]
感兴趣的基因组位置的列表的一些实例可见于表1和表2中。在一些实施方案中,本公开内容的方法中使用的基因组位置包括表1中的至少5个、至少10个、至少15个、至少20个、至少25个、至少30个、至少35个、至少40个、至少45个、至少50个、至少55个、至少60个、至少65个、至少70个、至少75个、至少80个、至少85个、至少90个、至少95个或97个基因中的至少一部分。在一些实施方案中,本公开内容的方法中使用的基因组位置包括表1中的至少5个、至少10个、至少15个、至少20个、至少25个、至少30个、至少35个、至少40个、至少45个、至少50个、至少55个、至少60个、至少65个或70个snv。在一些实施方案中,本公开内容的方法中使用的基因组位置包括表1中的至少1个、至少2个、至少3个、至少4个、至少5个、至少6个、至少7个、至少8个、至少9个、至少10个、至少11个、至少12个、至少13个、至少14个、至少15
个、至少16个、至少17个或18个cnv。在一些实施方案中,本公开内容的方法中使用的基因组位置包括表1中的至少1个、至少2个、至少3个、至少4个、至少5个或6个融合。在一些实施方案中,本公开内容的方法中使用的基因组位置包括表1中的至少1个、至少2个或3个插入或缺失中的至少一部分。在一些实施方案中,本公开内容的方法中使用的基因组位置包括表2中的至少5个、至少10个、至少15个、至少20个、至少25个、至少30个、至少35个、至少40个、至少45个、至少50个、至少55个、至少60个、至少65个、至少70个、至少75个、至少80个、至少85个、至少90个、至少95个、至少100个、至少105个、至少110个或115个基因中的至少一部分。在一些实施方案中,本公开内容的方法中使用的基因组位置包括表2中的至少5种、至少10种、至少15种、至少20种、至少25种、至少30种、至少35种、至少40种、至少45种、至少50种、至少55种、至少60种、至少65种、至少70种或73种snv。在一些实施方案中,本公开内容的方法中使用的基因组位置包括表2中的至少1个、至少2个、至少3个、至少4个、至少5个、至少6个、至少7个、至少8个、至少9个、至少10个、至少11个、至少12个、至少13个、至少14个、至少15个、至少16个、至少17个或18个cnv。在一些实施方案中,本公开内容的方法中使用的基因组位置包括表2中的至少1个、至少2个、至少3个、至少4个、至少5个或6个融合。在一些实施方案中,本公开内容的方法中使用的基因组位置包括表2中的至少1种、至少2种、至少3种、至少4种、至少5种、至少6种、至少7种、至少8种、至少9种、至少10种、至少11种、至少12种、至少13种、至少14种、至少15种、至少16种、至少17种或18种插入或缺失中的至少一部分。这些感兴趣的基因组位置中的每一个都可以被鉴定为给定诱饵集组的骨架区域或热点区域。感兴趣的热点基因组位置的列表的实例可见于表3中。在一些实施方案中,本公开内容的方法中使用的基因组位置包括表3中的至少1个、至少2个、至少3个、至少4个、至少5个、至少6个、至少7个、至少8个、至少9个、至少10个、至少11个、至少12个、至少13个、至少14个、至少15个、至少16个、至少17个、至少18个、至少19个、或至少20个基因中的至少一部分。列出了每个热点基因组位置的若干特征,包括相关基因、其所在的染色体、代表基因基因座的基因组的起始和终止位置、基因基因座以碱基对的长度、基因覆盖的外显子、以及可能寻求捕获的感兴趣的给定基因组位置的关键特征(例如,突变类型)。
[0186]
表1
[0187][0188]
表2
[0189][0190]
表3
[0191]
[0192]
[0193][0194]
在一些实施方案中,组中的一个或更多个区域包括来自一个或多于一个基因的一个或更多个基因座,用于检测手术之后残留的癌症。这种检测可以比现有癌症检测方法可能的检测更早。在一些实施方案中,组中的一个或更多个基因组位置包括来自一个或多于一个基因的一个或更多个基因座,用于检测高风险患者群体的癌症。例如,吸烟者患肺癌的比率比一般群体高得多。此外,吸烟者还可能发展其他肺部状况,这使癌症检测更加困难,诸如肺中不规则结节的发展。在一些实施方案中,本文描述的方法比现有的癌症检测方法可能的检测更早地检测高风险患者的癌症。
[0195]
可以选择待包括在测序组中的基因组位置,所述选择基于在该基因或区域中具有肿瘤标志物的患有癌症的受试者的数目。可以基于患有癌症的受试者的患病率和该基因中存在的肿瘤标志物来选择待包括在测序组中的基因组位置。区域中肿瘤标志物的存在可以指示受试者患有癌症。
[0196]
在一些情况下,可以使用来自一个或更多个数据库的信息来选择组。关于癌症的信息可以从癌症肿瘤活组织检查或cfdna测定获得。数据库可以包括描述被测序的肿瘤样品群体的信息。数据库可以包括关于肿瘤样品中mrna表达的信息。数据库可以包括关于肿瘤样品中调控元件或基因组区域的信息。与被测序的肿瘤样品相关的信息可以包括各种遗传变异的频率,并描述遗传变异发生的基因或区域。遗传变异可以是肿瘤标志物。这样的数据库的一个非限制性实例是cosmic。cosmic是在多种癌症中发现的体细胞突变的目录。对于特定的癌症,cosmic基于突变频率将基因排名。可以通过在给定基因中具有高突变频率来选择基因以包括在组中。例如,cosmic表明,33%的被测序的乳腺癌样品群体在tp53中具有突变,并且22%的被取样的乳腺癌群体在kras中具突变。其他被排名的基因,包括apc,具有仅见于约4%的被测序乳腺癌样品群体中的突变。基于在被取样的乳腺癌中具有相对高的频率(例如,与以约4%的频率存在的apc相比),tp53和kras可以被包括在测序组中。cosmic被作为非限制性实例提供,但是,可以使用将癌症与位于基因或遗传区域中的肿瘤标志物相关联的任何数据库或信息集。在另一个实例中,如由cosmic提供的,在1156个胆管癌样品中,380个样品(33%)在tp53中携带突变。其他若干基因,诸如apc,在所有样品中的4%-8%中具有突变。因此,可以基于在胆管癌样品群体中相对高的频率来选择tp53以包括在组中。
[0197]
可以为组选择肿瘤标志物在被取样的肿瘤组织或循环肿瘤dna中的频率显著高于在给定背景群体中发现的频率的基因或基因组区段。可以选择将多个基因组位置的组合包括在组中,使得至少大多数患有癌症的受试者可以在该组中的至少一个基因组位置或基因中存在肿瘤标志物或基因组区域。基因组位置的组合可以基于对于特定的癌症或一组癌症指示大多数受试者在所选择的区域的一个或更多个中具有一种或更多种肿瘤标志物的数据来选择。例如,为了检测癌症1,可以基于指示90%患有癌症1的受试者在组的区域a、b、c和/或d中具有肿瘤标志物的数据来选择包括区域a、b、c和/或d的组。可选地,可能显示肿瘤标志物独立地存在于患有癌症的受试者的两个或更多个区域中,使得两个或更多个区域组合中的肿瘤标志物存在于大多数患有癌症的受试者群体中。例如,为了检测癌症2,可以基于指示90%的受试者在一个或更多个区域中具有肿瘤标志物,并且在30%的这样的受试者中仅在区域x中检测到肿瘤标志物,而对于被检测出肿瘤标志物的其余受试者仅在区域y和/或z中检测出肿瘤标志物的数据来选择包括区域x、y和z的组。如果50%或更多时间在这些区域的一个或更多个中检测出肿瘤标志物,则在一个或更多个基因组位置中存在的先前显示与一种或更多种癌症相关的肿瘤标志物可以指示或预测受试者患有癌症。给定一个或更多个区域内的一组肿瘤标志物的癌症频率,可以使用计算方法诸如采用检测癌症的条件概率的模型来预测哪些区域单独或组合地可以预测癌症。用于组选择的其他方法包括使用数据库,所述数据库描述来自采用以大组和/或全基因组测序(wgs、rna-seq、芯片-seq、亚硫酸氢盐测序、atac序列及其他)进行的肿瘤综合基因组谱分析的研究的信息。从文献收集的信息也可以描述某些癌症中通常受影响及突变的途径。通过使用描述遗传信息的知识本体,可以进一步为组选择提供信息。
[0198]
被包括在测序组中的基因可以包括完全转录区、启动子区、增强子区、调控元件和/或下游序列。为了进一步增加检测指示肿瘤的突变的可能性,可以仅将外显子包括在组中。组可以包括选择的基因的所有外显子,或者仅包括选择的基因的一个或更多个外显子。组可以包括来自多于一个不同基因中每一个的外显子。组可以包括来自多于一个不同基因中每一个的至少一个外显子。
[0199]
在一些方面,选择来自多于一个不同基因中每一个的外显子的组,使得确定比例的患有癌症的受试者在外显子的组中的至少一个外显子中显示出遗传变异。
[0200]
可以对来自基因组中每一个不同基因中的至少一种完整外显子进行测序。被测序的组可以包括来自多于一个基因的外显子。组可以包括来自2个至100个不同基因、来自2个至70个基因、来自2个至50个基因、来自2个至30个基因、来自2个至15个基因、或来自2个至10个基因的外显子。
[0201]
选择的组可以包括不同数目的外显子。组可以包括从2个至3000个外显子。组可以包括从2个至1000个外显子。组可以包括从2个至500个外显子。组可以包括从2个至100个外显子。组可以包括从2个至50个外显子。组可以包括不多于300个外显子。组可以包括不多于200个外显子。组可以包括不多于100个外显子。组可以包括不多于50个外显子。组可以包括不多于40个外显子。组可以包括不多于30个外显子。组可以包括不多于25个外显子。组可以包括不多于20个外显子。组可以包括不多于15个外显子。组可以包括不多于10个外显子。组可以包括不多于9个外显子。组可以包括不多于8个外显子。组可以包括不多于7个外显子。
[0202]
组可以包括来自多于一个不同基因的一个或更多个外显子。组可以包括来自多于
一个不同基因的一部分中的每一个的一个或更多个外显子。组可以包括来自不同基因的每一个的至少25%、50%、75%或90%中的至少两个外显子。组可以包括来自不同基因的每一个的至少25%、50%、75%或90%中的至少三个外显子。组可以包括来自不同基因的每一个的至少25%、50%、75%或90%中的至少四个外显子。
[0203]
测序组的尺寸可能有所不同。测序组可以根据几个因素而变得更大或更小(就核苷酸尺寸而言),这些因素包括,例如,被测序的核苷酸总量或针对组中特定区域测序的独特分子的数目。测序组的尺寸可以是5kb至50kb。测序组的尺寸可以是10kb至30kb。测序组的尺寸可以是12kb至20kb。测序组的尺寸可以是12kb至60kb。测序组的尺寸可以是至少10kb、12kb、15kb、20kb、25kb、30kb、35kb、40kb、45kb、50kb、60kb、70kb、80kb、90kb、100kb、110kb、120kb、130kb、140kb或150kb。测序组的尺寸可以小于100kb、90kb、80kb、70kb、60kb或50kb。
[0204]
选择用于测序的组可以包括至少1个、5个、10个、15个、20个、25个、30个、40个、50个、60个、80个或100个基因组位置(例如,每个基因组位置包含感兴趣的基因组区域)。在一些情况下,选择组中的基因组位置,使得位置的尺寸相对较小。在一些情况下,组中的区域具有的尺寸为约10kb或更小、约8kb或更小、约6kb或更小、约5kb或更小、约4kb或更小、约3kb或更小、约2.5kb或更小、约2kb或更小、约1.5kb或更小、或约1kb或更小或者更小。在一些情况下,组中的基因组位置具有的尺寸为从约0.5kb至约10kb、从约0.5kb至约6kb、从约1kb至约11kb、从约1kb至约15kb、从约1kb至约20kb、从约0.1kb至约10kb或从约0.2kb至约1kb。例如,组中的区域可以具有的尺寸为从约0.1kb至约5kb。
[0205]
本文选择的组可以允许足以(例如,在从样品获得的无细胞核酸分子中)检测低频率遗传变异的深度测序。样品中遗传变异的量可以依据给定遗传变异的次要等位基因频率来表示。次要等位基因频率可以指在给定核酸群体诸如样品中次要等位基因(例如,不是最常见的等位基因)出现的频率。处于低次要等位基因频率的遗传变异在样品中存在的频率可能相对较低。在一些情况下,组允许检测处于至少0.0001%、0.001%、0.005%、0.01%、0.05%、0.1%或0.5%的次要等位基因频率的遗传变异。组可以允许检测处于0.001%或更高的次要等位基因频率的遗传变异。组可以允许检测处于0.01%或更高的次要等位基因频率的遗传变异。组可以允许检测样品中以低至0.0001%、0.001%、0.005%、0.01%、0.025%、0.05%、0.075%、0.1%、0.25%、0.5%、0.75%或1.0%的频率存在的遗传变异。组可以允许检测样品中以至少0.0001%、0.001%、0.005%、0.01%、0.025%、0.05%、0.075%、0.1%、0.25%、0.5%、0.75%或1.0%的频率存在的肿瘤标志物。组可以允许检测样品中处于低至1.0%的频率的肿瘤标志物。组可以允许检测样品中处于低至0.75%的频率的肿瘤标志物。组可以允许检测样品中处于低至0.5%的频率的肿瘤标志物。组可以允许检测样品中处于低至0.25%的频率的肿瘤标志物。组可以允许检测样品中处于低至0.1%的频率的肿瘤标志物。组可以允许检测样品中处于低至0.075%的频率的肿瘤标志物。组可以允许检测样品中处于低至0.05%的频率的肿瘤标志物。组可以允许检测样品中处于低至0.025%的频率的肿瘤标志物。组可以允许检测样品中处于低至0.01%的频率的肿瘤标志物。组可以允许检测样品中处于低至0.005%的频率的肿瘤标志物。组可以允许检测样品中处于低至0.001%的频率的肿瘤标志物。组可以允许检测样品中处于低至0.0001%的频率的肿瘤标志物。组可以允许在被测序的cfdna中检测样品中处于低至1.0%至0.0001%的频
率的肿瘤标志物。组可以允许在被测序的cfdna中检测样品中处于低至0.01%至0.0001%的频率的肿瘤标志物。
[0206]
在一定百分比的患有疾病(例如癌症)的受试者群体中可能显示出遗传变异。在一些情况下,患有癌症的群体的至少1%、2%、3%、5%、10%、20%、30%、40%、50%、60%、70%、80%、90%、95%或99%在组中的至少一个区域中显示出一种或更多种遗传变异。例如,患有癌症的群体的至少80%可以在组中的至少一个基因组位置中显示出一个或更多个遗传变异。
[0207]
组可以包括来自一个或更多个基因中的每一个的一个或更多个包含感兴趣的基因组区域的位置。在一些情况下,组可以包括来自至少1个、2个、3个、4个、5个、6个、7个、8个、9个、10个、15个、20个、25个、30个、40个、50个或80个基因中的每一个的一个或更多个包含感兴趣的基因组区域的位置。在一些情况下,组可以包括来自至多1个、2个、3个、4个、5个、6个、7个、8个、9个、10个、15个、20个、25个、30个、40个、50个或80个基因中的每一个的一个或更多个包含感兴趣的基因组区域的位置。在一些情况下,组可以包括来自从约1个至约80个、从1个至约50个、从约3个至约40个、从5个至约30个、从10个至约20个不同基因中的每一个的一个或更多个包含感兴趣的基因组区域的位置。
[0208]
可以选择该组中的区域,使得它们包含跨一个或更多个组织差异转录的序列。在一些情况下,包含基因组区域的位置可以包括在某些组织中相比于其他组织以较高水平转录的序列。例如,包含基因组区域的位置可以包括在某些组织中转录但在其他组织中不转录的序列。
[0209]
组中的基因组位置可以包括编码和/或非编码序列。例如,组中的基因组位置可以包括外显子、内含子、启动子、3’非翻译区、5’非翻译区、调控元件、转录起始位点和/或剪接位点中的一个或更多个序列。在一些情况下,组中的区域可以包括其他非编码序列,所述其他非编码序列包括假基因、重复序列、转座子、病毒元件和端粒。在一些情况下,组中的基因组位置可以包括非编码rna中的序列,所述非编码rna例如,核糖体rna、转移rna、piwi相互作用rna和微rna。
[0210]
可以选择组中的多个基因组位置以以期望的灵敏度水平检测(诊断)癌症(例如,通过检测一种或更多种遗传变异)。例如,可以选择组中的多个区域以以至少50%、55%、60%、65%、70%、75%、80%、85%、90%、95%、96%、97%、98%、99%、99.5%或99.9%的灵敏度检测癌症(例如,通过检测一种或更多种遗传变异)的区域。可以选择组中的多个基因组位置以以100%的灵敏度检测癌症。
[0211]
可以选择组中的多个基因组位置以以期望的特异性水平检测(诊断)癌症(例如,通过检测一种或更多种遗传变异)。例如,可以选择组中的多个基因组位置以以至少50%、55%、60%、65%、70%、75%、80%、85%、90%、95%、96%、97%、98%、99%、99.5%或99.9%的特异性检测癌症(例如,通过检测一种或更多种遗传变异)。可以选择组中的多个基因组位置以以100%的特异性检测一种或更多种遗传变异。
[0212]
可以选择组中的多个基因组位置以以期望的阳性预测值检测(诊断)癌症。阳性预测值可以通过增加灵敏度(例如,检测到实际阳性的几率)和/或特异性(例如,不将实际阴性误认为阳性的几率)来增加。作为非限制性实例,可以选择组中的多个基因组位置以以至少50%、55%、60%、65%、70%、75%、80%、85%、90%、95%、96%、97%、98%、99%、
99.5%或99.9%的阳性预测值检测一种或更多种遗传变异。可以选择组中的多个区域以以100%的阳性预测值检测一种或更多种遗传变异。
[0213]
可以选择组中的多个基因组位置以以期望的准确度检测(诊断)癌症。如本文使用的,术语“准确度”可以指测试区分疾病状况(例如,癌症)和健康状况的能力。准确度可以使用量度诸如灵敏度和特异性、预测值、似然比、roc曲线下面积、约登指数和/或诊断比值比来定量。
[0214]
准确度可以表示为百分比,是指给出正确结果的测试数和进行的测试总数之间的比率。可以选择组中的多个区域以至少50%、55%、60%、65%、70%、75%、80%、85%、90%、95%、96%、97%、98%、99%、99.5%或99.9%的准确度检测癌症。可以选择组中的多个基因组位置以以100%的准确度检测癌症。
[0215]
可以选择高度灵敏并检测低频率遗传变异的组。例如,可以选择组,使得可以以至少50%、55%、60%、65%、70%、75%、80%、85%、90%、95%、96%、97%、98%、99%、99.5%或99.9%的灵敏度检测样品中以低至0.01%、0.05%或0.001%的频率存在的遗传变异或肿瘤标志物。可以选择组中的基因组位置以以70%或更高的灵敏度检测样品中以1%或更低的频率存在的肿瘤标志物。可以选择组以以至少50%、55%、60%、65%、70%、75%、80%、85%、90%、95%、96%、97%、98%、99%、99.5%或99.9%的灵敏度检测样品中低至0.1%的频率的肿瘤标志物。可以选择组以以至少50%、55%、60%、65%、70%、75%、80%、85%、90%、95%、96%、97%、98%、99%、99.5%或99.9%的灵敏度检测样品中低至0.01%的频率的肿瘤标志物。可以选择组以以至少50%、55%、60%、65%、70%、75%、80%、85%、90%、95%、96%、97%、98%、99%、99.5%或99.9%的灵敏度检测样品中低至0.001%的频率的肿瘤标志物。
[0216]
可以选择高度特异性并检测低频率遗传变异的组。例如,可以选择组,使得可以以至少50%、55%、60%、65%、70%、75%、80%、85%、90%、95%、96%、97%、98%、99%、99.5%或99.9%的特异性检测样品中以低至0.01%、0.05%或0.001%的频率存在的遗传变异或肿瘤标志物。可以选择组中的基因组位置以以70%或更高的特异性检测样品中以1%或更低的频率存在的肿瘤标志物。可以选择组以以至少70%、75%、80%、85%、90%、95%、96%、97%、98%、99%、99.5%或99.9%的特异性检测样品中低至0.1%的频率的肿瘤标志物。可以选择组以以至少70%、75%、80%、85%、90%、95%、96%、97%、98%、99%、99.5%或99.9%的特异性检测样品中低至0.01%的频率的肿瘤标志物。可以选择组以以至少70%、75%、80%、85%、90%、95%、96%、97%、98%、99%、99.5%或99.9%的特异性检测样品中低至0.001%的频率的肿瘤标志物。
[0217]
可以选择高度准确并检测低频率遗传变异的组。可以选择组,使得可以以至少70%、75%、80%、85%、90%、95%、96%、97%、98%、99%、99.5%或99.9%的准确度检测样品中以低至0.01%、0.05%或0.001%的频率存在的遗传变异或肿瘤标志物。可以选择组中的基因组位置以以70%或更高的准确度检测样品中以1%或更低的频率存在的肿瘤标志物。可以选择组以以至少70%、75%、80%、85%、90%、95%、96%、97%、98%、99%、99.5%或99.9%的准确度检测样品中低至0.1%的频率的肿瘤标志物。可以选择组以以至少70%、75%、80%、85%、90%、95%、96%、97%、98%、99%、99.5%或99.9%的准确度检测样品中低至0.01%的频率的肿瘤标志物。可以选择组以以至少70%、75%、80%、85%、90%、95%、
96%、97%、98%、99%、99.5%或99.9%的准确度检测样品中低至0.001%的频率的肿瘤标志物。
[0218]
可以选择高度预测性并检测低频率遗传变异的组。可以选择组,使得样品中以低至0.01%、0.05%或0.001%的频率存在的遗传变异或肿瘤标志物可以具有至少70%、75%、80%、85%、90%、95%、96%、97%、98%、99%、99.5%或99.9%的阳性预测值。
[0219]
组中使用的探针或诱饵的浓度可以增加(2ng/μl至6ng/μl),以捕获样品中更多的核酸分子。组中使用的探针或诱饵的浓度可以为至少2ng/μl、3ng/μl、4ng/μl、5ng/μl、6ng/μl或更高。探针的浓度可以为约2ng/μl至约3ng/μl、约2ng/μl至约4ng/μl、约2ng/μl至约5ng/μl、约2ng/μl至约6ng/μl。组中使用的探针或诱饵的浓度可以为2ng/μl或更高至6ng/μl或更低。在一些情况下,这可以允许生物样品中的更多分子被分析,从而能够使较低频率的等位基因被检测到。
[0220]
b.癌症和其他疾病
[0221]
在某些实施方案中,本文公开的方法和方面用于诊断患者的特定疾病、紊乱或状况。通常,所考虑的疾病是癌症的类型。这样的癌症的非限制性实例包括胆道癌、膀胱癌、移行细胞癌、尿路上皮癌、脑癌、神经胶质瘤、星形细胞瘤、乳腺癌、化生癌、宫颈癌、宫颈鳞状细胞癌、直肠癌、结肠直肠癌、结肠癌、遗传性非息肉性结肠直肠癌、结肠腺癌、胃肠间质瘤(gist)、子宫内膜癌、子宫内膜间质肉瘤、食管癌、食管鳞状细胞癌、食管腺癌、眼黑素瘤、葡萄膜黑素瘤、胆囊癌、胆囊腺癌、肾细胞癌、透明细胞肾细胞癌(clear cell renal cell carcinoma)、移行细胞癌、尿路上皮癌、肾母细胞瘤、白血病、急性淋巴细胞白血病(all)、急性髓性白血病(aml)、慢性淋巴细胞白血病(cll)、慢性髓性白血病(cml)、慢性粒单核细胞白血病(cmml)、肝癌(liver cancer)、肝癌(liver carcinoma)、肝细胞瘤、肝细胞癌、胆管癌、肝母细胞瘤、肺癌、非小细胞肺癌(nsclc)、间皮瘤、b细胞淋巴瘤、非霍奇金淋巴瘤、弥漫性大b细胞淋巴瘤、套细胞淋巴瘤、t细胞淋巴瘤、非霍奇金淋巴瘤、前体t淋巴母细胞淋巴瘤/白血病、外周t细胞淋巴瘤、多发骨髓瘤、鼻咽癌(npc)、神经母细胞瘤、口咽癌、口腔鳞状细胞癌、骨肉瘤、卵巢癌、胰腺癌、胰腺导管腺癌、假乳头状肿瘤、泡细胞癌。前列腺癌、前列腺腺癌、皮肤癌、黑素瘤、恶性黑素瘤、皮肤黑素瘤、小肠癌、胃癌(stomach cancer)、胃癌(gastric carcinoma)、胃肠间质瘤(gist)、子宫癌、或子宫肉瘤。
[0222]
使用本文公开的方法和系统任选地评估的其它基于遗传的疾病、紊乱或状况的非限制性实例包括软骨发育不全、α-1抗胰蛋白酶缺乏症、抗磷脂综合征、孤独症、常染色体显性多囊肾病、夏科-马里-图思病(cmt)、猫叫综合征、克罗恩病、囊性纤维化、dercum病、唐氏综合征、duane综合征、杜兴氏肌营养不良症、因子v leiden易栓症、家族性高胆固醇血症、家族性地中海热、脆性x综合征、戈谢病、血色素沉着病、血友病、全前脑畸形、亨廷顿病、克兰费尔特综合征、马方综合征、强直性肌营养不良、神经纤维瘤病、努南综合征、成骨不全、帕金森病、苯丙酮尿症、poland异常、卟啉症、早老症、视网膜色素变性、重症联合免疫缺陷病(scid)、镰状细胞病、脊髓性肌萎缩症、泰-萨克斯病、地中海贫血、三甲基胺尿症、特纳综合征、颚心脸综合征(velocardiofacial syndrome)、wagr综合征、威尔逊病等。
[0223]
c.定制疗法和相关施用
[0224]
在一些实施方案中,本文公开的方法涉及对患有给定疾病、紊乱或状况的患者进行鉴定并施用疗法。基本上,任何癌症疗法(例如,手术疗法、放射疗法、化学疗法和/或类似
疗法)都被包括作为这些方法的一部分。通常,疗法包括至少一种免疫疗法(或免疫治疗剂)。免疫疗法通常指增强针对给定癌症类型的免疫应答的方法。在某些实施方案中,免疫疗法是指增强针对肿瘤或癌症的t细胞应答的方法。
[0225]
在一些实施方案中,免疫疗法或免疫治疗剂靶向免疫检查点分子。某些肿瘤能够通过利用(co-opting)免疫检查点途径来逃避免疫系统。因此,靶向免疫检查点已经成为对抗肿瘤逃避免疫系统的能力和活化针对某些癌症的抗肿瘤免疫的有效方法。pardoll,nature reviews cancer,2012,12:252-264.
[0226]
在某些实施方案中,免疫检查点分子是抑制性分子,其减少t细胞对抗原的应答中涉及的信号。例如,ctla4表达在t细胞上,并通过结合抗原呈递细胞上的cd80(又名b7.1)或cd86(又名b7.2)在下调t细胞活化中发挥作用。pd-1是另一种表达在t细胞上的抑制性检查点分子。pd-1在炎症反应期间限制外周组织中t细胞的活化。另外,在许多不同肿瘤的表面上pd-1的配体(pd-l1或pd-l2)通常被上调,导致肿瘤微环境中抗肿瘤免疫应答的下调。在某些实施方案中,抑制性免疫检查点分子是ctla4或pd-1。在其他实施方案中,抑制性免疫检查点分子是pd-1的配体,诸如pd-l1或pd-l2。在其他实施方案中,抑制性免疫检查点分子是ctla4的配体,诸如cd80或cd86。在其他实施方案中,抑制性免疫检查点分子是淋巴细胞活化基因3(lag3)、杀伤细胞免疫球蛋白样受体(kir)、t细胞膜蛋白3(tim3)、半乳糖凝集素9(gal9)或腺苷a2a受体(a2ar)。
[0227]
靶向这些免疫检查点分子的拮抗剂可用于增强针对某些癌症的抗原特异性t细胞应答。相应地,在某些实施方案中,免疫疗法或免疫治疗剂是抑制性免疫检查点分子的拮抗剂。在某些实施方案中,抑制性免疫检查点分子是pd-1。在某些实施方案中,抑制性免疫检查点分子是pd-l1。在某些实施方案中,抑制性免疫检查点分子的拮抗剂是抗体(例如,单克隆抗体)。在某些实施方案中,抗体或单克隆抗体是抗ctla4抗体、抗pd-1、抗pd-l1抗体或抗pd-l2抗体。在某些实施方案中,抗体是单克隆抗pd-1抗体。在一些实施方案中,抗体是单克隆抗pd-l1抗体。在某些实施方案中,单克隆抗体是抗ctla4抗体和抗pd-1抗体、抗ctla4抗体和抗pd-l1抗体、或抗pd-l1抗体和抗pd-1抗体的组合。在某些实施方案中,抗pd-1抗体是派姆单抗(pembrolizumab)或纳武单抗(nivolumab)中的一种或更多种。在某些实施方案中,抗ctla4抗体是伊匹单抗(ipilimumab)。在某些实施方案中,抗pd-l1抗体是阿特珠单抗阿维鲁单抗(avelumab)或德瓦鲁单抗(durvulumab)中的一种或更多种。
[0228]
在某些实施方案中,免疫疗法或免疫治疗剂是针对cd80、cd86、lag3、kir、tim3、gal9或a2ar的拮抗剂(例如抗体)。在其他实施方案中,拮抗剂是抑制性免疫检查点分子的可溶性形式,诸如包含抑制性免疫检查点分子的细胞外结构域和抗体的fc结构域的可溶性融合蛋白。在某些实施方案中,可溶性融合蛋白包含以下的细胞外结构域:ctla4、pd-1、pd-l1或pd-l2。在一些实施方案中,可溶性融合蛋白包含以下的细胞外结构域:cd80、cd86、lag3、kir、tim3、gal9或a2ar。在一种实施方案中,可溶性融合蛋白包含pd-l2的细胞外结构域或lag3的细胞外结构域。
[0229]
在某些实施方案中,免疫检查点分子是共刺激分子,其放大t细胞对抗原的应答中涉及的信号。例如,cd28是一种表达在t细胞上的共刺激受体。当t细胞通过其t细胞受体与
抗原结合时,cd28与抗原呈递细胞上的cd80(又名b7.1)或cd86(又名b7.2)结合,以放大t细胞受体信号传导并促进t细胞活化。因为cd28与ctla4结合相同的配体(cd80和cd86),所以ctla4能够抵消或调节通过cd28介导的共刺激信号传导。在某些实施方案中,免疫检查点分子是选自cd28、诱导性t细胞共刺激因子(icos)、cd137、ox40或cd27的共刺激分子。在其他实施方案中,免疫检查点分子是共刺激分子的配体,包括例如cd80、cd86、b7rp1、b7-h3、b7-h4、cd137l、ox40l或cd70。
[0230]
靶向这些共刺激检查点分子的激动剂可用于增强针对某些癌症的抗原特异性t细胞应答。相应地,在某些实施方案中,免疫疗法或免疫治疗剂是共刺激检查点分子的激动剂。在某些实施方案中,共刺激检查点分子的激动剂是激动剂抗体,且优选地是单克隆抗体。在某些实施方案中,激动剂抗体或单克隆抗体是抗cd28抗体。在其他实施方案中,激动剂抗体或单克隆抗体是抗icos抗体、抗cd137抗体、抗ox40抗体或抗cd27抗体。在其他实施方案中,激动剂抗体或单克隆抗体是抗cd80抗体、抗cd86抗体、抗b7rp1抗体、抗b7-h3抗体、抗b7-h4抗体、抗cd137l抗体、抗ox40l抗体或抗cd70抗体。
[0231]
除了癌症之外,用于治疗特定的基于基因的疾病、紊乱或状况的治疗选择通常是本领域普通技术人员熟知的,并且鉴于所考虑的特定疾病、紊乱或状况是明显的。
[0232]
在某些实施方案中,本文描述的定制疗法通常被肠胃外(例如,静脉内或皮下)施用。包含免疫治疗剂的药物组合物通常被静脉内施用。某些治疗剂是口服施用的。然而,定制疗法(例如,免疫治疗剂等)也可以通过本领域已知的任何方法施用,包括例如口服施用、舌下施用、直肠施用、阴道施用、尿道内施用、局部施用、眼内施用、鼻内施用和/或耳内施用,这些施用可以包括片剂、胶囊、颗粒、水性悬浮液、凝胶、喷雾剂、栓剂、药膏、油膏等。
实施例
[0233]
实施例1:检测ppg
[0234]
使用来自guardant healt,inc.(redwood city,ca)的73-基因的组的cfdna测试对17,825个临床样品的集合进行处理和分析。在该集合中,107个样品被鉴定为含有112个样品特异性ppg,如下表4中所示。这相当于每个样品的ppg比率为0.6%,或者每167个临床样品中有一个样品特异性ppg。
[0235]
表4
[0236][0237]
在表4中,示出了在至少1个样品中检测到样品特异性ppg的所有基因,而所有的singleton被组合在“singleton”类别中。
[0238]
由种系和体细胞样品特异性ppg产生的跨外显子-外显子接点的比对假象可能产生假的变异识别,如图5中所示。缺少内含子序列的多个软剪读段的存在以及内含子-外显
子边界处覆盖的不连续性揭示了ppg的存在。以1.7%的等位基因频率(af)观察到箭头指示的假的a.c snv识别。
[0239]
实施例2:ppg的临床结果
[0240]
ppg的存在可以导致两个不同来源的假阳性变异识别。首先,由ppg产生的跨ppg外显子-外显子接点的读段中的比对假象可以产生假的变异识别(图6)。其次,ppg中存在的snv可以映射到原始基因。
[0241]
使用其中未检测到ppg的10,000个临床样品的随机子集,观察到几个基因的ppg拷贝的存在导致在内含子-外显子边界处(图7a)和编码序列(cds)内(图7b)比本来预期的偶然snv更多的snv。
[0242]
实施例3:消除假阳性变异
[0243]
总之,剪接接点中的48个snv以及cds中的32个snv被确定为可能归因于源自hras、raf1、smad4和tp53的ppg的存在。通过对假阳性变异进行ppg知晓抑制(ppg-aware suppression),避免了每个样品0.45%(80/17,825)的假阳性率增加,如表5中所示。
[0244]
表5
[0245][0246]
实施例4:检测并抑制由tyro3 ppg引起的假阳性变异
[0247]
使用来自guardant healt,inc.(redwood city,ca)的500-基因的组的cfdna测试对2,094个患者样品的集合进行处理和分析。在该集合中,1,140个样品被鉴定为含有样品特异性ppg,例如基因tyro3。这相当于每个样品的ppg比率为54%,或者每两个样品有一个ppg。评估了这些样品的在15号染色体上位置41,862,477处的tyro3基因座的怀疑的假阳性的c>t突变(称为tyro3 c.1422c>t)的存在。
[0248]
表6
[0249][0250]
在表6中,在11个检测到ppg的样品中观察到怀疑的假阳性变异,但在没有检测到ppg的样品中没有观察到怀疑的假阳性变异,这是一个统计学上显著的差异(费希尔精确检验,p=0.0013)。由于该变异只在ppg存在的情况下观察到,这表明该变异是源自与tyro3基因座比对的ppg的读段的假象。
[0251]
由ppg产生的跨外显子-外显子接点的比对假象显示在tyro3基因座的背景中,如图8中所示。假的c.t.snv识别(tyro3 c.1422c>t)由箭头指示。
[0252]
虽然本文已经示出并描述了本公开内容的多种实施方案,但对于本领域技术人员将理解,这些实施方案仅通过实例的方式提供。在不偏离本公开内容的情况下,本领域技术人员可以想到许多变化、改变和替换。应当理解,可以采用本文描述的本公开内容的实施方案的多种替代方案。
[0253]
以上或以下引用的所有专利申请、网站、其他出版物、登录号等都为了所有目的被通过引用以其整体并入,其程度如同每个单独的项目都被具体且单独地指示通过引用如此并入一样。如果一个序列的不同版本在不同时间与一个登录号相关联,则意指在本申请的实际提交日期与该登录号相关联的版本。如果适用的话,有效提交日期意指真实提交日期或提及该登记号的优先权申请的提交日期中较早的一个。同样,如果出版物、网站等的不同版本在不同时间发布,则意指在本申请的实际提交日期最近发布的版本,除非另有指示。除非另外明确指出,否则本公开内容的任何特征、步骤、要素、实施方案或方面可以与任何其他结合使用。尽管为了清楚和理解的目的,已经通过说明和实例的方式在一些细节方面描述了本公开内容,但将明显的是,可以在所附权利要求书的范围内实施某些改变和修改。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1