一种借助高通量测序检测SMN1基因突变的方法和系统与流程
本发明属于基因检测和分析领域。具体而言,本发明涉及通过高通量测序和特殊的分析方法来检测smn1基因突变,特别是检测smn1基因第7外显子纯合缺失的装置、方法和系统。本发明还涉及利用本发明的装置、方法和系统来诊断脊髓性肌萎缩症(sma)或鉴别性诊断sma以及与sma表型易混淆的其他疾病,以及其上储存有本发明的方法的机器可读介质和设备。
背景技术:
脊髓性肌萎缩症(sma;omim#253300)是脑干和脊髓前角运动神经元丧失所导致的神经肌肉疾患,其为一种常染色体隐性遗传病,绝大多数患者由运动神经元生存1(smn1)基因的纯合缺失所导致。在中国人群中,携带sma相关smn1杂合缺失的概率约为1/42(sheng-yuan,z.等,molecularcharacterizationofsmncopynumberderivedfromcarrierscreeningandfromcorefamilieswithsmainachinesepopulation.eurjhumgenet,2010.18(9):第978-84页)。据最新统计,中国台湾地区sma致病基因携带率约1~3%,发病率约为1/17,000(chien,y.h.,etal.,presymptomaticdiagnosisofspinalmuscularatrophythroughnewbornscreening.jpediatr,2017),与中国大陆相似。流行病学调查发现,高达98%的sma患者以第5号染色体5q13区的运动神经元生存基因1(smn1)纯合缺失为遗传致病方式(sangare,m.等,geneticsoflowspinalmuscularatrophycarrierfrequencyinsub-saharanafrica.annneurol,2014.75(4):第525-32页;rad,i.a.,mutationspectrumofsurvivalmotorneurongeneinspinalmuscularatrophy.jdownsyndrchrabnorm,2017.3(1):第1-2页)。smn1基因在脊髓前角运动神经元轴突生长,神经肌肉接头突触形成等多种生理过程中起到了至关重要的作用(杨兰、宋昉,脊髓性肌萎缩症的治疗研究进展,《中华儿科杂志》,2016.54(8):第634-637页)。由于此基因缺陷导致的编码蛋白缺失关联到了多种跨系统疾患(singh,r.n.等,diverseroleofsurvivalmotorneuronprotein.biochimbiophysacta,2017.1860(3):第299-315页),同时也由于部分患者临床特征不典型,因而产生了临床鉴别诊断的需求。
临床上依据sma的特征可做出疑似诊断,具体的方法包括肌电图、肌肉活检组织化学染色以及血清肌酸磷酸激酶检测。但是鉴于sma在临床表现上个体差异较大,并且与多种其他疾病相似,缺乏特征性,前述这些检查手段或者不适用于婴幼儿患者,或者对检测条件要求较高,因此在确诊与鉴别性诊断上还需依赖于特殊分子检测。自steege与其同事在1995年将限制性片段长度多态性pcr(pcr-rflp)技术用于sma诊断以来,后续又出现了多重连接依赖探针扩增技术(mlpa)和实时荧光定量pcr(qpcr)方法学,能够在外显子水平识别出携带smn1/2缺失/重复的现象(arkblad,e.l.,etal.,multiplexligation-dependentprobeamplificationimprovesdiagnosticsinspinalmuscularatrophy.neuromusculdisord,2006.16(12):第830-8页)。但这些检测技术的诊断病种单一,在应用于诊断时,这些检测的结果只能揭示受检对象所患疾病“是否为sma”,但不能在结果为阴性时回答“若不是sma,应为何种疾病”的问题,因而限制了在鉴别性诊断sma及与之具有类似表型的疾病等方面的应用。
近年来,以全外显子组测序(wholeexomsequencing,wes)为代表的二代基因组测序技术(ngs)因其高通量和高性价比而被日益广泛地应用于遗传病诊断领域。由于wes的检测范围涵盖了人类基因组全部约两万个基因的编码区域,因而除能够检出致病性smn1纯合缺失外,也可有助于在病因学上鉴别那些与sma临床表型谱相近,而遗传病因不同的病例,从而在神经肌肉病的精准诊断上具备独特的优势。然而,人体中还存在smn基因的另一个着丝粒拷贝,即smn2,或称smn着丝粒型。由于转录时遗漏外显子7,smn2只编码非常少量的全长smn蛋白和大量的smn截短形式smnδ7。由于smn1与smn2基因高度同源,wes的常规数据分析方法难以鉴别两者,故被认为不适用于sma的分子诊断。
因此,本领域仍然需要一种性价比高、准确、全面的诊断方法,用于鉴定smn1的外显子7的纯合缺失以诊断sma,并且还能进一步鉴别sma和与之表型类似的疾病。
技术实现要素:
本发明提供了一种用于检测smn1基因突变,特别是作为sma最常见致病突变的smn1纯合缺失突变的装置、方法和系统,其借助高通量测序,并使用特殊算法分析高通量测序结果,能够针对所述smn1基因突变实现不亚于传统的“金标准”检测技术mlpa的高检出率及准确性。另外,本发明的装置、方法和系统由于采用了高通量测序技术,其测序结果中还可包含与其它神经肌肉疾病相关的基因的信息,进而不仅能用于诊断sma,还能对具有与sma相似的临床特征的其它神经肌肉病实现鉴别性诊断。本发明的方法和系统通过借助特殊算法,消除了本领域之前认为高通量测序如全外显子组测序不适于检测smn1纯合缺失突变时面临的问题,由此完成了本发明。
第一方面,本发明涉及一种用于检测受试者的smn1基因中纯合突变的分析装置,其中所述分析装置包括:
读取模块,其用于读取通过测序获得的信息,所述信息包括多个包含smn1基因的外显子7第840位点的reads;
计算模块,所述计算模块计算(smn1外显子7第840位点为碱基c的reads数)/(包含smn1外显子7第840位点的总reads数)的比值,
判定模块,当所述比值等于0或接近于0时,判定所述受试者为存在smn1外显子7的纯合缺失的阳性受试者,否则则判定所述受试者为不存在smn1外显子7的纯合缺失阴性受试者。
在第一方面的实施方案中,所述分析装置中的计算模块在执行所述计算之前,过滤掉平均质量值20以下的reads序列,优选过滤掉评价质量值25以下的reads序列。
在第一方面的实施方案中,所述分析装置中的计算模块在执行所述计算之前,过滤掉smn1外显子7第840位点的质量值小于10的reads。优选地,所述计算模块在执行所述计算之前,过滤掉smn1外显子7第840位点的质量值小于20的reads。更优选地,所述计算模块在执行所述计算之前,过滤掉smn1外显子7第840位点的质量值小于25的reads。
在第一方面的实施方案中,所述分析装置中的计算模块在执行所述计算之前,去除pcr扩增的重复序列。
在第一方面的实施方案中,所述接近于0的比值是小于0.1的比值。
在第一方面的实施方案中,所述的分析装置用于sma的诊断。在进一步的实施方案中,所述sma是在遗传上与smn的突变相关的。在更进一步的实施方案中,所述sma选自sma-i型、sma-ii型、sma-iii型和sma-iv型。进一步或可选地,所述分析装置用于sma与其他同sma具有类似表型的疾病的鉴别性诊断。在优选的实施方案中,所述同sma具有类似表型的疾病为神经肌肉疾病。
第二方面,本发明涉及用于检测受试者的smn1基因中纯合突变的系统,其中所述系统包括:
测序装置,所述测序装置对多个扩增子进行测序,所述多个扩增子通过扩增来自受试者的样品中的核酸获得并且包含smn1基因的外显子7第840位点,所述测序产生多个包含smn1基因的外显子7第840位点的reads;和第一方面所述的分析装置。
在一个实施方案中,本发明的测序是高通量测序。在优选的实施方案中,本发明使用的高通量测序选自下组:对smn1基因的测序、对smn1外显子7的测序、包含smn1基因或其第7外显子的panel测序、全基因组测序(wholegenomesequencing,wgs)、全外显子组测序(wholeexomsequencing,wes)或临床外显子组测序(clinicalexomsequencing,ces)。在更优选的实施方案中,本发明的高通量测序是全外显子组测序或临床外显子组测序。
在一个实施方案中,本发明的分析装置读取的信息中smn1外显子7第840位点的reads数为10至100万个。例如,本发明的分析装置读取的信息中的reads数为至少10个,至少50个,至少100个,至少1000个,至少10000个,至少10万个或至少100万个。
在进一步的实施方案中,所述系统还包括扩增装置,所述扩增装置对来自所述受试者的包含核酸的样本进行扩增以产生多个包含smn1基因的外显子7第840位点的扩增子,将所述多个扩增子用于所述测序装置。
在一个实施方案中,本发明的分析装置或系统用于诊断脊髓性肌萎缩症(sma)。在进一步的实施方案中,所述sma是在遗传上与smn的突变相关的。在更进一步的实施方案中,所述sma选自sma-i型、sma-ii型、sma-iii型和sma-iv型。进一步或可选地,所述分析装置或系统用于sma与其他同sma具有类似表型的疾病的鉴别性诊断。在优选的实施方案中,所述同sma具有类似表型的疾病为神经肌肉疾病。
在一个实施方案中,本发明的分析装置或系统用于鉴别性诊断sma和与sma表型相类似的疾病。在具体的实施方案中,所述与sma表型相类似的疾病选自:becker型肌营养不良症、bethlem肌病、kleefstra综合征、merosin缺乏性先天性肌肉萎缩症、ullrich型先天性肌营养不良、x连锁肌管性肌病、x连锁中央核肌病、ywhae基因miller-dieker综合征、先天性糖基化病1a型(omim:212065)、先天性肌无力综合征4a型、先天性肌病(早发,伴心肌病)、巨颅伴皮层下海绵样囊中个性脑白质病1型、常染色体显性下肢遗传脊髓性肌萎缩症、常染色体隐性肌硬化症、常染色体隐性遗传远端型脊髓型肌萎缩2型(omim:605726)、杜氏肌营养不良/进行性假肥大性肌营养不良症、肌萎缩厕所硬化症(asl)16型(omim:614373)、肢带型肌营养不良症2j型(omim:608807)、胼胝体发育不全伴周围神经病变、遗传性肌病伴早起呼吸衰竭和遗传性运动感觉性神经病vi型。
在一个实施方案中,本发明所述分析装置中的模块之间可以通过有线连接和无线连接。
第三方面,本发明涉及一种用于检测受试者的smn1基因中纯合突变的方法,所述方法包括:
(1)读取来自测序的信息,所述信息包括多个包含smn1基因的外显子7第840位点的reads;
(2)计算(smn1外显子7第840位点为碱基c的reads数)/(包含smn1外显子7第840位点的总reads数)的比值;和
(3)当所述比值等于0或接近于0时,判定所述受试者为存在smn1外显子7的纯合缺失的阳性受试者,否则将所述受试者判定为不存在smn1外显子7的纯合缺失阴性受试者。
第四方面,本发明涉及一种机器可读的介质,其包含机器可读代码,所述代码在由机器实施时执行如下操作以检测受试者的smn1基因中纯合突变的存在:
(1)读取来自测序的信息,所述信息包括多个包含smn1基因的外显子7第840位点的reads;
(2)计算(smn1外显子7第840位点为碱基c的reads数)/(包含smn1外显子7第840位点的总reads数)的比值;和
(3)当所述比值等于0或接近于0时,判定所述受试者为存在smn1外显子7的纯合缺失的阳性受试者,否则将所述受试者判定为不存在smn1外显子7的纯合缺失阴性受试者。
在第三和第四方面的实施方案中,在执行第(2)步的计算之前,过滤掉smn1外显子7第840位点的质量值小于10的reads。优选地,在执行所述计算之前,过滤掉smn1外显子7第840位点的质量值小于20的reads。更优选地,在执行所述计算之前,过滤掉smn1外显子7第840位点的质量值小于25的reads。
在第三和第四方面的实施方案中,在执行第(2)步的计算之前,去除pcr扩增的重复序列。
在第三和第四方面的实施方案中,所述接近于0的比值是小于0.1的比值。
在第三和第四方面的实施方案中,所述方法或所述机器可读的介质用于sma的诊断。在进一步的实施方案中,所述sma是在遗传上与smn的突变相关的。在更进一步的实施方案中,所述sma选自sma-i型、sma-ii型、sma-iii型和sma-iv型。进一步或可选地,所述方法或所述机器可读的介质用于sma与其他同sma具有类似表型的疾病的鉴别性诊断。在优选的实施方案中,所述同sma具有类似表型的疾病为神经肌肉疾病。在具体的实施方案中,所述与sma具有类似表型的疾病选自:becker型肌营养不良症、bethlem肌病、kleefstra综合征、merosin缺乏性先天性肌肉萎缩症、ullrich型先天性肌营养不良、x连锁肌管性肌病、x连锁中央核肌病、ywhae基因miller-dieker综合征、先天性糖基化病1a型(omim:212065)、先天性肌无力综合征4a型、先天性肌病(早发,伴心肌病)、巨颅伴皮层下海绵样囊中个性脑白质病1型、常染色体显性下肢遗传脊髓性肌萎缩症、常染色体隐性肌硬化症、常染色体隐性遗传远端型脊髓型肌萎缩2型(omim:605726)、杜氏肌营养不良/进行性假肥大性肌营养不良症、肌萎缩厕所硬化症(asl)16型(omim:614373)、肢带型肌营养不良症2j型(omim:608807)、胼胝体发育不全伴周围神经病变、遗传性肌病伴早起呼吸衰竭和遗传性运动感觉性神经病vi型。
在第三和第四方面的实施方案中,所述来自测序的信息中smn1外显子7第840位点的reads数为10至100万个,例如至少10个,至少50个,至少100个,至少1000个,至少10000个,至少10万个或至少100万个。所述测序是高通量测序。在优选的实施方案中,本发明使用的高通量测序选自下组:对smn1基因的测序、对smn1外显子7的测序、包含smn1基因或其第7外显子的panel测序、全基因组测序(wgs)、全外显子组测序(wes)或临床外显子组测序(ces)。在更优选的实施方案中,本发明的高通量测序是全外显子组测序或临床外显子组测序。
第五方面,本发明涉及一种设备,其包含本发明第四方面所述的机器可读的介质。
第六方面,本发明涉及本发明的第一方面所述的分析装置、第二方面所述的系统、第四方面的介质和第五方面的设备用于诊断sma的用途或鉴别性诊断sma与其他同sma具有类似表型的疾病的用途。
附图说明
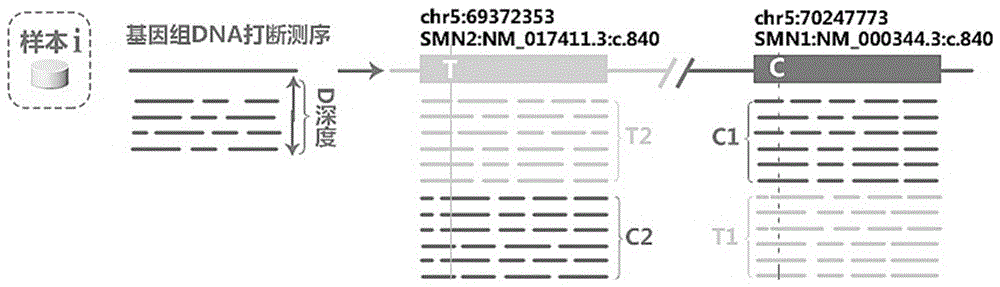
图1示例性显示了将样本经历基因组测序之后,通过比对算法burrows-wheeler将获得的reads序列随机分配到smn1和smn2的处理方式。
图2的表格列出了将本发明用于鉴别性诊断时,在通过本发明诊断为非smn1纯合缺失的受试者中,为21名受试者进行了进一步诊断,并确诊为为其他神经肌肉疾病。图2中列出了所述21名受试者的年龄、突变类型和诊断出的疾病。
发明详述
除非另有说明,否则本文公开的一些方法的实践采用免疫学,生物化学,化学,分子生物学,微生物学,细胞生物学,基因组学和重组dna的常规技术,这些技术在本领域的技术范围内。参见例如sambrook和green,molecularcloning:alaboratorymanual,4thedition(2012);系列分子生物学(f.m.ausubel,etal.eds.);系列方法在酶学(academicpress,inc。),pcr2:apracticalapproach(m.j.machersrs,b.d.hamesandg.r.tayloreds.(1995)),harlowandlane,eds.(1988)antibodies,alaboratorymanual,andcultureofanimalcells:amanualofbasictechniqueandspecializedapplications,6thedition(r.i.breshney,ed.(2010))。
术语“约”或“近似”意指在本领域普通技术人员确定的特定值的可接受误差范围内,这将部分取决于如何测量或确定该值,即,测量系统的局限性。例如,根据本领域的实践,“约”可以表示在1或大于1的标准偏差内。或者,“约”可表示给定值的最多20%,最多10%,最多5%或最多1%的范围。或者,特别是对于生物系统或过程,该术语可以表示数值的一个数量级,优选地在5倍内,更优选地在2倍内。在申请和权利要求中描述特定值的情况下,除非另有说明,否则应当假定术语“约”意味着在特定值的可接受误差范围内。
术语“受试者”、“个体”或“患者”在本发明的上下文中可以互换使用,指脊椎动物,优选哺乳动物,例如啮齿类动物、灵长类动物,更优选人。
如本文所用的术语“基因”是指核酸(例如dna,例如基因组dna和cdna)及其相应的编码rna转录物的核苷酸序列。如本文所用,关于基因组dna的术语包括插入的非编码区以及调节区,并且可包括5'和3'末端。在一些用途中,该术语包括转录序列,包括5'和3'非翻译区(5'-utr和3'-utr),外显子和内含子。在一些基因中,转录区域将包含编码多肽的“开放阅读框”。在该术语的一些用途中,“基因”仅包含编码多肽所必需的编码序列(例如,“开放阅读框”或“编码区”)。在一些情况下,基因不编码多肽,例如核糖体rna基因(rrna)和转移rna(trna)基因。在一些情况下,术语“基因”不仅包括转录序列,而且还包括非转录区域,包括上游和下游调节区,增强子和启动子。基因可以指生物基因组中其天然位置中的“内源基因”或天然基因。基因可以指“外源基因”或非天然基因。非天然基因可以指通常不在宿主生物体中发现但通过基因转移引入宿主生物体的基因。非天然基因也可以指不在生物体基因组中的天然位置的基因。非天然基因还可以指天然存在的核酸或多肽序列,其包含突变,插入和/或缺失(例如,非天然序列)。
如本文所用的术语“核苷酸”通常是指碱-糖-磷酸盐组合。核苷酸可包含合成核苷酸。核苷酸可包含合成的核苷酸类似物。核苷酸可以是核酸序列的单体单元(例如脱氧核糖核酸(dna)和核糖核酸(rna))。术语核苷酸可包括核糖核苷三磷酸腺苷三磷酸(atp),尿苷三磷酸(utp),三磷酸胞嘧啶(ctp),三磷酸鸟苷(gtp)和脱氧核糖核苷三磷酸如datp,dctp,ditp,dutp,dgtp,dttp或其衍生物。这些衍生物可包括,例如,[αs]datp,7-脱氮-dgtp和7-脱氮-datp,以及赋予含有它们的核酸分子核酸酶抗性的核苷酸衍生物。本文使用的术语核苷酸可以指双脱氧核糖核苷三磷酸(ddntp)及其衍生物。双脱氧核糖核苷三磷酸的说明性实例可包括但不限于ddatp,ddctp,ddgtp,dditp和ddttp。核苷酸可以通过众所周知的技术进行未标记或可检测标记。标记也可以用量子点进行。可检测标记可包括例如放射性同位素,荧光标记,化学发光标记,生物发光标记和酶标记。
术语“多核苷酸”,“寡核苷酸”和“核酸”可互换使用,指任何长度的聚合形式的核苷酸,脱氧核糖核苷酸或核糖核苷酸,或其类似物,可以是单链、双链或多链形式。多核苷酸对细胞可以是外源的或内源的。多核苷酸可以存在于无细胞环境中。多核苷酸可以是其基因或片段。多核苷酸可以是dna。多核苷酸可以是rna。多核苷酸可以具有任何三维结构,并且可以执行已知或未知的任何功能。多核苷酸可包含一种或多种类似物(例如改变的主链,糖或核碱基)。
本文所述“脊肌萎缩症”,或称脊髓性肌萎缩症(sma),其主要的致病基因为smn1。基于目前医学遗传学的共识,sma主要与两个高度同源的基因密切相关,smn1与smn2,这两个基因主要通过7号外显子和8号外显子上的两个基因位点进行区分(quyjetal,pmid:27425821,jmoldiagn,2016)。smn1和smn2有五个碱基对的差异,但是这五个碱基差异都不会导致氨基酸序列的变化,但是由于其中在smn2外显子7中的一个c到t的变化影响了外显子的剪接,导致从smn2获得的全长转录物大幅减少。大部分正常个体都有2份拷贝的smn1基因与2份拷贝的smn2基因。smn2基因由于发生了外显子7的跳跃,只能产生少量的全长smnmrna,导致其补偿作用有限。因此,如果某个个体两份拷贝的smn1基因都失去功能,也就是出现smn1的纯合缺失时,该个体必然会患上sma疾病。与之相对,杂合型缺失smn1的个体同尚有一份正常的smn1等位基因,因此其不会展现出sma疾病的表型,将这样的个体称为smn1缺失突变的“携带者”。
本文所述smn1基因,与同源基因smn2之间的差别之一在于c.840位点的碱基差别(smn1为c,smn2为t),该位点处于外显子7中,因此也同时体现为外显子7的差别(smn1的外显子7是以c.840位点是碱基c为特征,而smn2的外显子7是以c.840位点是碱基t为特征)。若smn1基因的外显子7上的c.840位点的碱基c变异为t,则使得smn1的基因序列变为smn2的序列,故这种点突变的变异可理解为等同于不存在smn1的外显子7的正常拷贝。
在2018年5月中国国家卫生健康委员会、科技部、工业和信息化部、国家药品监督管理局、国家中医药管理局等五部门联合发布的第一批罕见病目录中,脊髓性肌萎缩症位列121种疾病之一。sma发病时间早,是一种儿科神经退行性疾病,严重者一般很难存活超过2岁。广义上的sma还包括一些与smn基因不相关且更为罕见的类型。因此,在本发明的上下文中,“sma”指由smn基因的纯合缺失点突变导致的类型,除非另行具体指明。临床上根据发病时间和严重程度,将与5号染色体上smn基因内的纯合缺失突变相关的sma进一步分为i型、ii型、iii型和iv型这四种亚型,各个亚型的临床表现存在差异。总的来说,发病越早的亚型越为严重。也有观点认为smn2的拷贝数可能对sma的严重程度有影响(
sma-i型又称werdnighoffman病,是最严重的亚型,其发病时间早,部分病例甚至早在胎儿期就会出现胎动减弱、变少的情况。其他病例也会在出生后数个月内发病,并在发病后一年内因呼吸衰竭死亡,一般无法存活超过2岁。sma-i型患者的临床表现包括对称性肌无力,大运动减少,6个月后不能独坐;肌肉弛缓,腱反射减低或消失;肌肉萎缩,但由于婴儿脂肪多而不易被发现;肋间肌麻痹;运动脑神经受损等。sma-ii型也称中间型或慢性sma,发病比sma-i型稍迟,通常在7月龄至1岁半发病,进展相对i型更为缓慢。临床表现包括严重的肌无力,患儿大多可以独坐但无法独立行走。sma-iii型也称为kugelberg-welander病,是儿科sma中发病最晚、表现最轻的类别。症状包括肌无力、肌肉萎缩等。sma-iv型为成年发病型sma,通常在二十岁至四十岁之间特别是三十几岁时确诊,sma-iv型患者通常具有4至6个smn2基因拷贝,可以部分弥补因smn1纯合缺失导致的smn蛋白的缺乏问题。
综合来看,本发明所述的sma的临床表现包括例如肌无力、肌张力低、肌肉弛缓、肌肉萎缩、肌麻痹、脊柱侧凸和弯曲,以及因此导致的行动不便、呼吸问题、进食和吞咽问题、大运动发育迟缓等。另外,sma患者还会在肌电图检查中表现出异常,展现出去神经支配现象。这些状况在临床上可以帮助sma的诊断和识别,但是由于存在其他症状和表现类似的疾病,通常在临床上还是需要分子学的检测来确诊sma。本发明的检测手段运用了高通量测序的结果,因此同时可以包含与其他遗传病相关的分子诊断信息,特别是与sma具有类似临床表现的其他遗传病相关的分子诊断信息。利用这一点就可以同时诊断受试者是否患有sma和这些其他遗传病,在确认患者所患疾病不是sma的情况下,仍能够为疾病确诊提供相关信息,做到“鉴别性诊断”。因此在本发明的上下文中,“鉴别性诊断”意指确定患者所患疾病,并排除其他与之具有类似的症状、临床表现的疾病的诊断。
本文所述高通量测序也称为大规模平行测序、下一代测序(nextgenerationsequencing,ngs),其特征在于可以对基因组的同一位置得到多个非重复片段的测序reads,从而提高测序结果数据的深度。在本发明的上下文中,“高通量测序”、“二代测序”、“下一代测序”、“ngs”可以互换使用。二代测序通常被认为包括使用焦磷酸测序法和dna聚合酶的454测序、使用边合成边测序法和dna聚合酶的solexa测序、使用连接酶测序法和dna连接酶的solid测序、使用半导体测序法和dna聚合酶的iontorrent测序等测序方法。在本发明的上下文中,所述高通量测序法指能够实现深度测序的测序方法,其能够获得样品有关基因组的特定位点的多个测序reads。如本领域通常理解的,“reads”指高通量测序过程中由每个反应产生的序列,通过对这些序列的读取形成测序的原始数据。通过对相互重叠的reads的拼接能够获得重叠群(contig),这一过程通常由测序拼接软件来完成。通过对重叠群的分析可以进一步匹配其中的重叠部分,并确定重叠群在基因组中的顺序,由顺序已知的重叠群组成的更长的scaffold。在能够实现获得多重reads这一测序结果的前提下,本发明的高通量测序不限于具体的测序原理、方法、仪器和/或试剂。在一个实施方案中,本发明所述的读取模块中的信息是这种由reads构成的原始数据。在一个实施方案中,所述由reads构成的原始数据通过本发明的系统中包含的测序装置获得。
在关于测序特别是高通量测序或称二代测序的上下文中,为每个碱基赋予质量值(q)来描述测序结果的准确度。例如,某个碱基的质量值为q20的含义是在碱基识别(basecalling)的过程中,对该碱基的识别结果给出的错误率为10的负2次方,即错误率为1%而正确率为99%,质量值为q30的含义是错误率为0.1%而正确率为99.9%,以此类推,因此质量值越高代表该碱基被错误测序的概率越小。q20大于等于90%的含义是,对于一定量的测序数据而言,其中90%的碱基数据的质量值能够达到q20或更好。具体到本发明中,就测序的整体质量而言,q20大于等于90%,并且q30大于等于85%。“平均质量值”是指就整个基因组所包括的碱基位置而言的整体平均质量值。
对于本发明的分析装置而言,所述计算模块在执行所述计算之前,过滤掉平均质量值20以下的reads序列,优选过滤掉平均质量值25以下的reads序列,更优选过滤掉平均质量值30以下的reads序列。可选地或者除此之外,所述计算模块在执行所述计算之前,优选过滤掉原始数据中smn1外显子7第840位点的质量值不到20的reads。这种过滤意味着去除了就smn1外显子7第840位点而言测序的正确率低于99%的reads。
可选地或者除此之外,所述计算模块在执行所述计算之前,去除pcr扩增的重复序列。
在关于测序特别是高通量测序或称二代测序的上下文中,“覆盖度(coverage)”指将测序结果组装后获得的基因组序列的大小占整个基因组大小的比例。测序中往往无法获得覆盖100%的基因组序列的测序结果,这是由基因组的固有组成和测序方法的不足所导致的,比如基因组中含有一些高gc含量区域、重复序列等复杂结构。
在关于测序特别是高通量测序或称二代测序的上下文中,“深度(depth)”或“测序深度”指测序中测序得到的总碱基数与待测基因组大小的比值。举例来说,测序深度为10x意味着获得的总数据量是整个基因组的十倍,基因组中的每个单一碱基被平均测序或读取了10次。在本发明的实施方案中,就测序的整体而言,深度在50x以上,优选60x以上,更优选70x以上,甚至更优选80x以上,甚至还更优选90x以上,还更优选100x以上。在具体的事实方案中,“深度”也意味着测序结果中包含特定位点的reads数。因此在具体的一个实施方案中,所述测序的单样本测序深度在80x以上。
因此,本发明所述的测序获得的信息中的10x覆盖度大于85%是指就被测序的整体序列而言,有85%的区域获得了至少10x的覆盖度。在本发明的具体实施方案中,所述测序获得的信息的10x的覆盖度大于85%,优选大于90%,更优选大于95%。
另外,在具体讨论smn1基因第7外显子区域的测序深度时,测序深度意味着包含感兴趣的smn1基因第7外显子区域840位点的reads数。如本文所述对包含smn1基因第7外显子区域的高通量测序,是可以借助这种测序,得到smn1基因第7外显子区域的深度测序数据,即可以得到该区域的多个非重复片段的测序reads数据,该区域的总reads数一般不小于10x,不小于15x、不小于20x、不小于30x、不小于40x、不小于50x、不小于60x、不小于70x、不小于80x、不小于90x、不小于100x。在优选的实施方案中,所述区域的测序平均q20≥90%,q30≥85%。
本文所述smn1的第7外显子纯合缺失,可以体现为包括但不限于三种情况:1)纯合缺失,即在smn1基因所在的染色体基因座的绝对坐标位置上,smn1基因两个等位基因的第7外显子拷贝数均缺失;2)纯合点突变,smn1基因的两个等位基因的外显子7上的c.840位点的碱基c变异为t,等同于smn1的两个等位基因的外显子7均缺失(即纯合缺失);3)缺失点突变杂合体,在smn1基因所在的染色体基因座的绝对坐标位置上,smn1基因一个等位基因的第7外显子拷贝数缺失,并且另一个等位基因的外显子7上的c.840位点的碱基c变异为t。这三种情况均视同于smn1的第7外显子的纯合缺失,即不存在正常的smn1拷贝。
本文所述的smn1的第7外显子的纯合缺失,无论是前述三种情况的哪一种,ngs数据都会体现为smn1基因的c.840位点体现为碱基c的缺失,该位置的碱基全部为碱基t,故该位置为碱基c的reads数与该位置为任意碱基的reads数(即该位置为c和t的reads数之和)之比(以下或表示为“r”其等于(smn1外显子7第840位点为碱基c的reads数)/(包含smn1外显子7第840位点的总reads数)为0。
除此之外,考虑到高通量测序存在技术局限性,本文的检测手段还将所述比值r接近于0的结果也判定为smn1纯合缺失阳性。具体而言,高通量测序存在一定概率的测序错误,比如将smn1基因的c.840位点的t错误的检测为c,则会造成r事实上本应为0但根据测序数据得出的结果却不为0的情况。随着测序技术的发展,目前测序错误的概率较低,比如illumima的hiseq系列或novaseq系列测序仪的测序错误概率大约为千分之一。因此,即使出现罕见的测序错误,如果针对c.840位点的总reads数越大,就会使得计算比值r时的分母越大,相应地错误对r值的影响就会越小,即便r本应为0但不为0,但其数值仍接近于0。在本发明的上下文中,接近于0具体可以是选自下组的数值,例如小于等于0.05即5%,小于等于0.03即3%,小于等于0.02或2%,小于等于0.01或1%,小于等于0.005或0.5%,小于等于0.003或0.3%,小于等于0.002或0.2%,甚至小于等于0.001或0.1%。具体来说,在测序使用illumima的hiseq系列或novaseq系列测序仪或碱基错误率相当的测序仪完成的情况下,将c/c+t接近于0的情况定义为小于等于0.1或10%,小于等于0.05或5%,小于等于0.01或1%,小于等于0.005或0.5%,小于等于0.003或0.3%,小于等于0.002或0.2%,甚至小于等于0.001或0.1%,在这些情况下,也将检测结果判定为smn1纯合缺失阳性。因此,本发明的r值接近于0也可以意味着其是一个小于等于测序体系系统误差的值。
本发明的方法中通过计算比值而非smn1的c.840位点为c的绝对reads数是否为零来预测拷贝数,其优势在于避免了因检测过程中产生的错误,如测序错误等而导致的非零情况归为smn1不为纯合缺失的情况,从而导致漏检。举例来说,若以smn1的c.840位点为c的reads数是否为零来判断是否为纯合缺失,那么在测序错误率为千分之一的情况下,smn1的c.840位点每测序1000x就会出现一次测序错误,假定测序错误正好为c时,当该位点测序1万x时,c可能出现10x,当该位点测序为10万x时,c可能出现100x,测序错误会导致c的绝对reads数较大,但是利用r=c/总reads数,则可以发现此时r的比值非常低,甚至接近于测序错误的系统误差,因此应该通过r来判断纯合缺失,而不应该使用c的绝对reads数。
本文所述包含smn1基因或其第7外显子的panel测序,是指对包含smn1基因或其第7外显子的一个以上的基因的组合(即panel)进行测序。
本文所述术语“全基因组测序(wholegenomesequencing,wgs)”,或“全外显子组测序(wholeexomsequencing,wes)”均按照本领域通常的理解来解读。其中,全外显子测序(wes)策略是一种诊断遗传病常用的ngs策略,其利用探针捕获并富集外显子区域的dna序列,再进行高通量测序发现与蛋白质变异相关的基因突变。外显子组测序的对象是基因组中的蛋白质编码区域,这些区域只占全基因组的不到2%,因而外显子组测序相较于基因组测序而言降低了实验和分析成本,可以实现更低的价格、更短的测序时间以及更深的覆盖度。
本文所述临床外显子组测序(clinicalexomsequencing,ces),或称医学外显子组测序,是指将多个已知的致病基因进行测序的策略。
本文所述mlpa,是指多重连接依赖探针扩增技术(multiplexligation-dependentprobeamplification,mlpa),mlpa技术最早是由荷兰学者dr.schouten于2002年提出的一种针对待测核酸中靶序列进行定性和定量分析的检测技术。原理是利用简单的探针和靶序列dna进行杂交,之后通过连接、pcr扩增,产物通过毛细管电泳分离及数据收集,最后利用软件对收集的数据进行分析得出结论的一种技术。是一种在同一反应管内检测多达50种核苷酸序列拷贝数变化的方法。该技术可以同时鉴定几十个基因或位点的缺失和插入。它是一种灵敏的技术,可以快速有效地定量核酸序列。它在世界各地的许多实验室进行,可用于检测基因的拷贝数变化(如缺失或复制),识别dna的甲基化状态,检测单核苷酸多态性(snps)和点突变,量化mrna。因此,它被应用于许多研究和诊断领域,如细胞遗传学、癌症研究、人类遗传学等。该技术曾是用于检测smn1第7外显子拷贝数的主流方法。
本发明的采样装置、扩增装置、测序装置、分析装置可以整合在一起,也可以为物理上各自独立的装置。当它们在物理上处于各自独立的状态下时,对于这些装置之间的距离没有限制,只要这些装置能够实现其在本发明的系统或方法中承担的功能即可。
用于本发明的测序的受试者样本基因组dna的制备可以通过本领域技术人员公知的方法和/或试剂盒来进行。受试者样本可以是体液、细胞、组织等,优选是血液。
本发明的方法和系统还可以用于鉴别性诊断。“鉴别性诊断”在本文的上下文中意指在多种疾病具有相似的临床表现形式的情况下,诊断并确定受试者所患疾病为所述多种疾病中的哪一种疾病。可以通过本发明的方法和系统与sma进行鉴别性诊断的疾病是那些可以通过基因测序诊断的遗传性疾病,其与sma具有一种或多种相似的临床表现,所述临床表现包括但不限于肌无力、肌张力低、肌肉弛缓、肌肉萎缩、肌麻痹,以及因此导致的行动不便、呼吸问题、进食和吞咽问题、大运动发育迟缓、脊柱侧凸和弯曲等。。在一些优选的实施方案中,所述与sma具有相似临床表现形式的疾病通常也为运动神经元疾病,特别是下肢运动神经元疾病。具体的实例包括但不限于becker型肌营养不良症、bethlem肌病、kleefstra综合征、merosin缺乏性先天性肌肉萎缩症、ullrich型先天性肌营养不良、x连锁肌管性肌病、x连锁中央核肌病、ywhae基因miller-dieker综合征、先天性糖基化病1a型(omim:212065)、先天性肌无力综合征4a型、先天性肌病(早发,伴心肌病)、巨颅伴皮层下海绵样囊中个性脑白质病1型、常染色体显性下肢遗传脊髓性肌萎缩症、常染色体隐性肌硬化症、常染色体隐性遗传远端型脊髓型肌萎缩2型(omim:605726)、杜氏肌营养不良/进行性假肥大性肌营养不良症、肌萎缩厕所硬化症(asl)16型(omim:614373)、肢带型肌营养不良症2j型(omim:608807)、胼胝体发育不全伴周围神经病变、遗传性肌病伴早起呼吸衰竭和遗传性运动感觉性神经病vi型。
具体实施方式
为了更全面地理解和应用本发明,下文将参考实施例和附图详细描述本发明,所述实施例仅是意图举例说明本发明,而不是意图限制本发明的范围。本发明的范围由后附的权利要求具体限定。
在本发明的实施例中采用了一种诊断遗传病常用的ngs策略——全外显子测序(wes)策略,并结合本发明所述的ngs数据分析方法,用于检测smn1基因是否存在第七外显子纯合缺失(实施例1),以及其他与sma表型相近的神经肌肉病的基因是否存在致病突变,进而用于鉴别性诊断(实施例2)。在下文中将这种方法统称为全外显子测序或wes。另外,为了验证使用本发明的方法和系统借助高通量测序(具体来说wes)和数据分析检出sma的准确性(accuracy),还对临床拟诊为sma的患者样本以sma诊断的“金标准”方法mlpa进行了诊断,将mlpa的诊断结果做为诊断参照,与采用本发明的方法和系统获得的结果进行比对(实施例3)。
实施例1.在临床拟诊为sma的受试者中检测smn1纯合缺失
本实施例涉及使用本发明的检测体系在临床拟诊为sma的受试者中检测smn1纯合缺失。
受试者的选择
本实施例的受试者为2015年6月至2018年7月期间在医院就诊的患者,于就诊期间从这些患者处取得了外周全血生物样本。入组病例均具有神经肌肉病特征性表型,由送检医师提供对应于每位患者的临床特征描述及各项特殊检查结果(患者的姓名等隐私信息已被隐藏)。对于样本检测结果将用于临床科研及数据发表的用途,在研究开始前已获得患者或监护人以及参加研究的家系成员的书面知情同意。
本实施例的受试者如表1中所示,240名受试者均在临床上被拟诊为sma,其中男性140名(58.3%),女性100名(41.7%),绝大多数受试者为儿童。
dna的提取和全外显子组(wes)测序
从来自患者的血液样品获得了基因组dna(dna要求浓度大于50ng/ul,总量达1μg),将获得的基因组dna通过超声进行破碎,在两端连接接头(illumina,sandiego,ca),加上标示样本的index序列,pcr扩增之后和生物素标记的探针杂交捕获目标序列。使用nimblegenseqcapezv2enrichmentkit(47mbp)富集芯片和seqcapezchoicekits(捕获最大7mbp的定制区域,包含smn1和smn2基因)进行dna捕获。采用illuminahiseq2500高通量测序仪进行测序。全外显子测序上机过程中,保证单样本测序深度(测序总数据量base个数/上述定制区域长度)80x以上,测序平均q20≥90%,q30≥85%,pe+se百分比≥95%,且10x以上覆盖度≥95%。数据分析使用碱基识别方法(callingmethod),对变体进行注释(annotate)。
使用本发明的算法检测smn1纯合缺失
smn1和smn2基因是高度相似的同源基因,共有5个碱基的差异,其中1个位于外显子7中,1个位于外显子8中,另外三个位于内含子中,而外显子7中含有终止密码子,外显子8不编码氨基酸,因此两者编码区仅有1个碱基的差异,即外显子7中的差异。具体来说,smn1基因染色体坐标chr5:70247773(nm_000344.3:c.840)为c,smn2基因染色体坐标位置上chr5:69372353(nm_017411.3:c.840)为t。
全外显子组测序范围不包含内含子区域,因此本发明的算法利用该单一位点进行拷贝数计算。考虑到所使用的短序列比对算法来自burrows-wheeler比对软件(alignersoftware),其为一种容忍错配的比对算法,因此会将实际上来自smn1和smn2的reads序列随机比对分配给这两个基因(如图1所示)。在图1中,smn2下的reads片段代表了被算法分配到smn2的reads数或深度;smn1下的reads片段代表了被算法分配到smn1的reads数或深度。但由于这是在容忍错配的情况下进行的随机分配,因此smn2之下实际上包含真正应为smn2的reads数或深度,将其称为t2,和实际为c但被错误地分配到smn2基因上的reads数或深度,将其称为c2。被分配到smn1基因的reads也存在类似的情况,其中实际上包含真正为c的reads数或深度,将其称为c1,和实际为t但被错误地分配到smn1基因上的reads数或深度,将其称为t1。因而需要计算两个位点上reads序列的深度,所有真正为c的reads数或称深度(c=c1+c2),以及真正为t的reads数或称深度(t=t1+t2)。由于c的检测值和smn1实际存在的拷贝数成正比,且t的检测值和smn2实际存在的拷贝数成正比,认为可以通过c:(c+t)的比例r来推算smn1是否是纯合缺失。当smn1发生纯合缺失时c碱基测序深度为0,则c/(c+t)比值r为0;如果smn2也同时表现为纯合缺失,则c=t=0。
考虑到建库、捕获、pcr、测序各个步骤都可能会引入碱基错误,数据统计是在对不可靠的reads序列进行了过滤后进行的。过滤的标准包括:过滤掉原始数据平均质量值20以下的reads序列,通过samtools软件去除pcr扩增的重复序列,过滤掉c.840位点碱基测序质量q20以下的reads,最终得到支持c与t的reads。另外,为了避免因系统误差导致的漏检,将算法设定c:(c+t)<0.1或者c去重复深度<3作为smn1纯合缺失(sma阳性)的判定阈值,否则判定为无smn1纯合缺失(sma阴性)。
根据上述方法获得的数据诊断数据如表1所示,在240名受试者中,诊断为smn1纯合缺失的受试者共122名。
本申请实施例的全部计数资料采用统计软件spss16.0,用成组t检验方法检验统计学显著性意义,p<0.05定义为有显著性统计学意义。
表1
*ns:不显著
实施例2.对非smn1纯合缺失的受试者的鉴别性诊断
采用全外显子检测遗传病的通用方法分析受试者(特别是在实施例1中鉴定为非smn1纯合缺失的118名受试者)是否具有其他与sma表型相近的神经肌肉疾病,以实现鉴别性诊断。
所述通用方法的具体步骤包括:
1)原始数据产量统计:去接头污染,过滤掉平均质量值低于20的reads,从reads末端过滤掉质量值低于20的碱基。
2)比对:数据与参考序列比对统计(比对软件bwa),参考基因组采用hg19基因组。
3)变异检测:用gatk对比对结果进行比对重排和质量矫正,然后使用gatk的haplotypecaller算法call突变。
4)突变假阳性过滤:根据测序深度、突变质量,对检测得到的单核苷酸变异(snv)、插入缺失(indel)进行过滤筛选,得到高质量可靠的突变:突变深度至少达到2x,突变率>10%,突变质量值>20的突变。
5)突变注释:根据snv和indel在基因上的位置,分析得到氨基酸变化影响,剪切影响,utr,内含子突变影响等。
6)筛选出的变异对蛋白功能影响的预测:利用provean,sift,polyphen2_hdiv,polyphen2_hvar,mutationtaster,m-cap,revel危害性预测软件基于同源比对,蛋白结构的保守性等的算法,预测筛选出的变异对蛋白质的影响。
7)使用maxentscan软件对剪切位点附近的突变做剪切危害性预测。
8)关联dbsnp,1000genome突变频率,exac数据库,omim,swiss-var数据库,注释已报道的疾病基因和已报道的致病位点,注释已报道突变的maf等。
9)按照2015年acmg国际指南进行遗传变异分级,筛选1-3级pathogenic/likelypathogenic/vus变异,结合变异所在的基因和关联的omim疾病遗传方式进行遗传判定,筛选出遗传模式支持致病的变异。
10)将受试者的临床表型与遗传模式支持的omim疾病表型进行匹配,找到和患者表型匹配的疾病做为候选疾病,并结合经治临床医师的判断得出最终分子诊断结论。
通过上述方法,将实施例1中确定为非smn1纯合缺失的118名患者中的21名受试者诊断为其他神经肌肉疾病,具体的诊断结果参见图2中的表格。换言之,对于这21名受试者而言,他们在最初的拟诊中都存在误诊(参见表1)。
实施例3.在临床为怀疑为sma的受试者中检测smn1纯合缺失
截至在2018年8月为止,发明人在全部具有神经肌肉病临床特征、但初步诊断未怀疑sma的受试者中,对于不能排除遗传致病因素的受试者,通过如实施例1中所述的本发明的检测手段检测出56例受试者携带smn1纯合缺失突变(参见表2)。
表2
*ns:不显著
综合实施例1至3中获得的结果,可以看出本发明的方法可以针对不同情况的受试者给出以下几项综合性诊断信息。
1)对临床初步判断为sma的240名患者进行了鉴别诊断:
a.122例确诊sma(122/240,50.8%):在实施例1中全部240例临床初步判断为sma的患者中,122例患者经wes检测smn1纯合突变阳性,即wes发现smn1基因外显子7纯合缺失,并且mlpa验证结果均为阳性,即smn1基因0拷贝;
b.22例避免了误判为sma,并确诊为其他疾病(22/240,9.2%):全部240例临床初步判断为sma的患者中,22例患者经wes检测smn1纯合突变阴性,即wes发现并非smn1基因外显子7的纯合缺失,并且mlpa验证结果均为阴性,即smn1基因大于0拷贝,并且wes检出其它导致患者神经肌肉疾患的相关致病性基因变异(具体情况参见图2);
c.96例排除了smn1第7外显子纯合缺失型sma(96/24040.0%):全部240例临床初步判断为sma的患者中,96例患者经wes检测smn1纯合突变阴性,即wes发现并非smn1基因外显子7的纯合缺失,并且mlpa阴性验证结果均为阴性,即smn1基因大于0拷贝,且wes未检出其它导致患者神经肌肉疾患的致病变异。
2)对临床未考虑sma、但实际是sma的患者,避免了漏诊sma
d.避免遗漏sma诊断:具有神经肌肉病临床表型,在接受检测前临床上未被怀疑为sma,经wes检出smn1纯合缺失,并由随后的mlpa验证所证实的携带者共56名。
实施例4.用mlpa方法对本发明的检测结果进行验证
为了验证本发明的方法的准确性,发明人对所有受试者样本均采用了现有技术中的金标准检测技术多重连接依赖探针扩增(mlpa)进行了验证,并将其结果与通过本发明的检测方法获得的结果进行了比较。
多重连接依赖性探针扩增用于检测smn1/smn2拷贝数变异,作为wes检测结果的验证手段。每次实验采用取自3名健康人的血样作为对照,其年龄及性别分布与入组受试者比较的统计学检验无显著性差异。mlpa试剂盒采用荷兰mrc-holland公司p060产品,包含30对探针,可以特异性检测smn1与smn2基因第7和第8号外显子的拷贝数(其中smn1第7外显子由于决定了此基因功能的完整性,故其拷贝数等同于等位基因的数目);该试剂盒中的4种探针检测smn1或smn2基因序列(表3),其他的探针均用来检测其他染色体作为参照。特异性检测smn1基因的外显子7探针位于183nt位置,其检测到的杂合缺失即表明sma携带。特异性检测smn1基因外显子8的探针位于218nt位置,可检测到95%外显子7拷贝数的变化(仅检测到smn1基因外显子8缺失不代表sma携带)。此外试剂盒包括了检测smn2基因外显子7(282nt)和外显子8(301nt)的探针和17对内对照物探针。
具体的实验流程如下。
1)杂交:取5μldna(终浓度为30ng/μl)加入ep管,98℃变性5min,冷却至25℃后滴加1.50μl多重探针及1.50μlbuffer,95℃变性1min后在60℃杂交16-24hrs。
2)连接:滴加32μl连接混合液,54℃孵育15min,98℃灭活连接酶5min。
3)扩增:取连接后的产物10μl,加入4μlpcrbuffer及26μlddh2o,72℃下加入10μl扩增反应液并启动pcr反应。反应条件为95℃变性30s,60℃退火30s,72℃延伸1min,共35个循环,最后72℃延伸20min。
4)分离:取1μl扩增产物加入8.7μlhi-di甲酰胺(美国abi公司)及0.30μlliz-500marker(美国abi公司)95℃变性5min,采用geneticanalyzer-3130基因分析仪(美国abi公司)进行毛细管电泳分离。
mlpa数据分析如下进行。采用genemapper3.0程序分析毛细管电泳分离结果,并导出图型及数据。将各目的片段峰面积除以全部内参照峰面积之和,即为该目的片段的相对峰面积(rpa),再将sma组rpa与正常对照组平均rpa(即20个正常对照rpa的平均值)相比较而得出拷贝数比值,进而可计算出该目的片段的拷贝数。根据荷兰mrc-holland公司官方网站(http://www.mlpa.com)提供的拷贝数定义标准,拷贝数比值范围在0.40-0.65为1拷贝,0.80-1.20为2拷贝,1.30-1.65为3拷贝,1.75-2.15为4拷贝。若某一片段无峰信号即代表该片段缺失。当拷贝数比值临近波动范围边界时实施重复验证以确保结果准确无误。
表3.mlpa引物序列
通过使用如实施例1中所述的本发明的算法分析wes测序结果,在实施例1和3涉及的296名患者中,发现了178例smn1纯合缺失(具体数值参见表5),118例非smn1纯合缺失。与通过上述mlpa检测获得的结果进行比对或发现,这些结果均与mlpa获得的结果一致(表4),符合率达100%,说明本发明的算法对smn1纯合缺失的诊断准确度、灵敏度和特异性均与现有技术中的金标准mlpa技术相当。
表4
另外,如实施例3中所总结的,本发明的方法能够获得综合性的诊断结果,特别是给出了mlpa无法提供的鉴别性诊断,因而体现出了比mlpa更强大的诊断能力。
表5.纯合缺失受试者的r值
实施例5.受试者的临床特征分析
根据本发明的研究对象的数据,可以按照最终的诊断结果(如实施例3中所示分为四组,即“确诊”、“误诊”、“病因未明”、“漏诊”)对受试者进行分组,归纳了每个组别的临床特征。
确诊受试者:小婴儿表现为哭声低,呼吸困难,和呼吸衰竭;其余患者全部具有对称性四肢肌无力(多数以近端与下肢为主),肌张力减弱,和相应的运动功能受损;部分患者还有肌颤,神经反射减弱,和肢体肌萎缩等临床特征。
误诊受试者:全部患者均具有肢体肌力减弱的共同特征。其余的临床表型则非sma所特有,而可归属于其它致病性基因变异所关联疾患的特征性表型谱,如假肥大性肌营养不良(也称duchenne型肌营养不良症或dmd)患者所具有的假性肌肥大与肌张力增高,以及其它基因突变所关联的面部畸形,发育延迟等。
病因未明的受试者:多数存在肢体肌力减弱,此外为非特异性临床特征,包括痫性发作,步态异常,发育延迟,肢体肌张力增高或减退,以及脑影像学异常发现等。
漏诊的受试者:小婴儿表现为哭声低和呼吸困难;其余全部患者均表现为肢体肌力减弱,肌张力正常或减低,但无增高现象,做过脑影像学检查者均未见明显异常。
以上各组病例,经治医师能够保持联系的病例在经由wes/mlpa检测明确病因诊断后,间隔6-12个月接受了病情随访,均证实了其临床特征未发生足以影响最终诊断疾病种类的变化。
从以上对临床特征的统计可见,各组受试者之间存在一些相同或类似的临床特征,进一步说明了需要本发明的方法,即一种比通过临床特征诊断更加准确,但又比mlpa更加全面的诊断方法。
序列表
<110>北京智因东方转化医学研究中心有限公司
<120>一种借助高通量测序检测smn1基因突变的方法和系统
<130>pq12731ctm33cn
<141>2020-04-08
<160>8
<170>siposequencelisting1.0
<210>1
<211>12
<212>dna
<213>人工序列(artificialsequence)
<400>1
ttacagggtttc12
<210>2
<211>12
<212>dna
<213>人工序列(artificialsequence)
<400>2
agacaaaatcaa12
<210>3
<211>12
<212>dna
<213>人工序列(artificialsequence)
<400>3
gtaaaagactgg12
<210>4
<211>12
<212>dna
<213>人工序列(artificialsequence)
<400>4
ggtgggggtggg12
<210>5
<211>12
<212>dna
<213>人工序列(artificialsequence)
<400>5
ttacagggtttt12
<210>6
<211>12
<212>dna
<213>人工序列(artificialsequence)
<400>6
agacaaaatcaa12
<210>7
<211>12
<212>dna
<213>人工序列(artificialsequence)
<400>7
gtaaaagactga12
<210>8
<211>12
<212>dna
<213>人工序列(artificialsequence)
<400>8
ggtgggggtggg12
- 还没有人留言评论。精彩留言会获得点赞!