一种包含沉降抑制剂的阿帕替尼口服药物组合物的制作方法
1.本公开属于制药领域,涉及一种包含阿帕替尼或其可药用盐的药物组合物,组合物中还包含沉降抑制剂。
背景技术:
2.生物药物分类系统中(bcs)ii类药物具有良好的渗透性和较差的溶解性。对这些药物来说,溶解度是吸收的限制。该类别中的弱碱性药物,也称为bcs iib药物,能够在酸性胃液中形成高浓度。ph值从胃到肠的自然转变会自动产生过饱和。但是过饱和是热力学不稳定的,它将很快沉淀。沉降抑制剂可以延长过饱和状态,并且可能持续足够长的时间以提高生物利用度,但是不同的沉降抑制剂对每种活性药物成分(api)表现出不同的亲和力。
3.筛选和评估沉淀抑制剂对过饱和的作用是有效开发过饱和药物递送系统的关键。多数研究都是通过反复试验筛选出一种适合药物的沉淀抑制剂。yamashita等通过基于96孔板的酶标仪技术采用高通量筛选方法选择沉淀抑制剂(yamashita,t.,ozaki,s.,kushida,i.,2011.int j pharm 419,170-174.)。price等通过计算药物-聚合物混合焓,开发了一种新颖的筛选方案,用于选择过饱和制剂的沉淀抑制剂(price,d.j.,nair,a.,kuentz,m.,dressman,j.,saal,c.,2019.european journal of pharmaceutical sciences 132,142-156.)。但是生理条件非常复杂,这些简单的体外测试通常在体内结果上会遇到矛盾(pestieau,a.,evrard,b.,2017.european journal of pharmaceutical sciences 102,203-219.)。
4.阿帕替尼是一种bcs ii类弱碱性药物(pka=4.72;log p=4.51(gastroplus预测);mw=493.58(甲磺酸阿帕替尼),397.48(阿帕替尼碱)),它是一种口服生物利用的小分子抑制剂,可选择性抑制血管内皮生长因子受体2。中阿帕替尼的口服摄入生物利用度在10%至20%之间(geng,r.,&li,j.(2015).expert opinion on pharmacotherapy,16(1),117-122.)。
技术实现要素:
5.本公开提供了一种包含阿帕替尼或其可药用盐的口服药物组合物,组合物中还包含沉降抑制剂。
6.在一些实施方案中,阿帕替尼或其可药用盐不是纳米晶形式。在一些实施方案中,阿帕替尼或其可药用盐的粒径d90值大于10μm。在一些实施方案中,当沉降抑制剂为羟丙甲基纤维素时,阿帕替尼或其可药用盐的d90大于10μm。
7.在一些实施方案中,所述的沉降抑制剂是聚合物、表面活性剂和环糊精等。
8.聚合物可选自:纤维素衍生物,例如hpmc、hpmcas、甲基纤维素(methyl cellulose,mc)、乙基纤维素(ethyl cellulose,ec)以及羟丙基纤维素(hydroxypropyl cellulose,hpc)等;乙烯基聚合物,例如pvp、聚乙烯醇(polyvinylalcohol,pva)以及聚乙烯吡咯烷酮共聚物(polyvinylpyrrolidone copolymer,copvp)等;环氧乙烷聚合物,例如
peg等。一些常见的表面活性剂有聚乙二醇-15-羟基硬脂酸酯(polyethyleneglycol-15-hydroxystearate,solutol hs 15),十二烷基硫酸钠(sodium dodecyl sulfate,sds)和tpgs等。
9.在一些实施方案中,所述的沉降抑制剂是羟丙甲基纤维素和/或共聚维酮。
10.在一些实施方案中,沉降抑制剂与阿帕替尼或其可药用盐的重量比可选自50:1-1:10,优选30:1-1:10,更优选20:1-1:10,更优选20:1-1:1,更优选更优选15:1
--
1:1、10:1-1:1、9:1-1:1、8:1-1:1、7:1-1:1、6:1-1:1、5:1-1:1、4:1-1:1、3:1-1:1、2:1-1:1。
11.本公开提供的药物组合物,组合物呈选自片剂、胶囊、微型片剂、颗粒剂或粉末剂的剂型。在一些实施方案中,组合物呈片剂。
12.本公开所述的药物组合物,还可以包含崩解剂,崩解剂可选自交联羧甲基纤维素钠、交联聚维酮、羧甲基淀粉钠、羧甲基纤维素钙、低取代羟丙基纤维素、淀粉、预胶化淀粉、海藻酸中的至少一种。
13.本公开所述的药物组合物,还可以包含填充剂,填充剂可选自微晶纤维素、乳糖、甘露醇、预胶化淀粉、糊精、山梨醇、蔗糖、磷酸氢钙、无水磷酸氢钙、硫酸钙等。
14.本公开所述的药物组合物,还可以包含粘合剂,粘合剂可选自预胶化淀粉、聚维酮、羟丙基纤维素、羧甲基纤维素钠、聚乙二醇等。
15.本公开所述的药物组合物,还可以包含润滑剂,润滑剂可选自硬脂酸、硬脂酸镁、微粉硅胶、滑石粉、聚乙二醇4000、聚乙二醇6000、棕榈酸、硬脂酸钙、胶态二氧化硅、巴西棕榈蜡、硬脂富马酸钠等。
16.在一些实施方案中,本公开所述的药物组合物中,阿帕替尼或其可药用盐的日剂量为50至700mg,优选300至600mg,更优选400至550mg,最优选500mg。
17.本公开还提供沉降抑制剂在制备包含阿帕替尼或其可药用盐的药物组合物中的用途。
18.本公开还提供一种双相胃肠模拟系统,包括3个容器1、2、3,分别模拟胃室、十二指肠腔室和回肠腔室,模拟胃室的容器1与模拟十二指肠腔室的容器2通过管状物相连通,模拟十二指肠腔室的容器2与模拟回肠腔室的容器3通过管状物相连通,还包括另外3个容器4、5、6,装有模拟胃液或模拟肠液,装有模拟胃液的容器4与模拟胃室通过管状物相连通,装有模拟肠液的容器5与模拟十二指肠腔室通过管状物相连通,装有模拟肠液的容器6与模拟回肠腔室通过管状物相连通,每个管状物上均设置蠕动泵,其中模拟十二指肠腔室中和模拟回肠腔室添加了正辛醇作为吸收相以模拟肠壁对肠道中药物的吸收,模拟胃室、十二指肠腔室和回肠腔室的容器中应用了pion在线光纤紫外检测系统。
19.在一些实施方案中,容器1、2、3均为溶出杯。
20.在一些实施方案中,模拟胃室、十二指肠腔室和回肠腔室的容器中还可以应用ph计。
21.在一些实施方案中,模拟胃室、十二指肠腔室和回肠腔室的容器均在37℃水浴中。
22.本公开还提供预测沉降抑制剂体内性能的方法,包括使用双相胃肠模拟系统。
23.本公开还提供一种预估口服药物在体内行为的方法,包括使用双相胃肠模拟系统。
24.本公开还提供预测沉降抑制剂用于阿帕替尼药物组合物中的体内性能的方法,包
括使用双相胃肠模拟系统。
附图说明
25.图1:双相胃肠模拟系统(bgis)示意图
26.图2:溶解度-ph图
27.图3:双相胃肠模拟系统各腔室药物浓度监测结果
28.图4a:十二指肠室过饱和度变化
29.图4b:空肠室过饱和度变化
30.图5:阿帕替尼沉淀过程显微图
31.图6:阿帕替尼沉淀物电镜图
32.图7:阿帕替尼沉淀的dsc热分析图
33.图8:阿帕替尼沉淀物的x射线衍射图
34.图9:阿帕替尼各组药时曲线
35.图10:体外溶出-时间曲线
36.图11:体内吸收分数-时间曲线
37.图12:levy图
38.图13:体外溶解分数与体内吸收分数之间的相关性
39.图14:预测和实测血药浓度-时间曲线
具体实施方式
40.实施例1:溶解度测定
41.溶出介质制备:
42.磷酸氢二钠
–
柠檬酸缓冲液
43.配制300ml 0.2mol/l na2hpo4和0.1mol/l柠檬酸溶液,加入标准fassif粉末,取100ml加入1000mg copvp,另取100ml加入1000mg hpmc,得到三组缓冲溶液(即无沉降抑制剂组,copvp组和hpmc组),各组分别以不同比例的混合配制ph值为2、3、4、4.7、5、6、6.5的磷酸氢二钠
–
柠檬酸缓冲溶液。
44.饱和溶解度样品制备:
45.取少量甲磺酸阿帕替尼,分别加入上述各缓冲液3ml(n=3),超声20min使api分散利于溶解,37℃震摇24h,4500rpm离心,取上清液200μl,与800μl流动相稀释液混匀,进样检测含量。
46.hplc含量测定:
47.标准品配制:取阿帕替尼游离碱25.00mg于25ml容量瓶,乙腈定容,分别稀释为浓度1、3、10、30、100、300、1000μg/ml.
48.色谱条件:以10mmol/l kh2po4(10%h3po4调ph至3.0)为流动相a,以乙腈为流动相b,流动相a:流动相b=60:40,流速1.0ml/min,等度洗脱10min,柱温30℃,检测波长260nm,进样量10μl,色谱柱kromasil 100-5c18(250
×
4.6mm,5um)。
49.结果如图2所示,正方形
■
表示没有pi(沉降抑制剂),三角形
▲
表示copvp,圆圈
●
表示hpmc。实线是拟合曲线。
50.实施例2:基于双相胃肠模拟系统的溶出检测
51.装置如附图1所示,有3个溶出杯分别代表胃、十二指肠、空肠;5个蠕动泵模拟药液的胃肠转运以及胃液、肠液的分泌;在生理相关介质水液上方的正辛醇作为吸收相,以模拟肠壁对肠道内药物的吸收;在胃腔室以及十二指腔室和回肠腔室的两相溶液中分别置入pion在线光纤紫外检测系统的探头,以实时监测各腔室药物浓度;此外在十二指肠腔室和回肠腔室的水相中置入ph探头,以监测胃肠转运过程中ph值的变化。
52.具体运行参数:
53.胃室模拟胃,初始为300ml含药fassgf,a泵模拟胃排空,将其泵入十二指肠室,泵速随时间t变化为v
out
=ln(2)*300*e^(-ln(2)*t/8)/8+1,c泵模拟胃液分泌泵入1ml/min fassgf;
54.十二指肠室模拟十二指肠,初始为50ml模拟肠液和100ml正辛醇,接受由a泵转运的fassgf,b泵将水相泵入回肠室,泵速随时间t变化为v=ln(2)*300*e^(-ln(2)*t/8)/8+2,同时蠕动泵d模拟肠液分泌泵入v=1ml/min;
55.空肠室模拟空肠,初始为100ml模拟肠液和100ml正辛醇,接受由b泵转运的模拟肠液,同时e泵模拟肠液分泌泵入v=1ml/min。
56.a、b两泵通过由arduino软件编程的单片机控制,使胃腔室内液体排空满足一级动力学速度,同时满足十二指肠腔室体积恒定。各泵运转时间为40min,此后各腔室溶液体积保持恒定。
57.各腔室均在37℃水浴中,以磁力搅拌100rpm。
58.各腔室水相体积v随时间t变化可由下式描述:
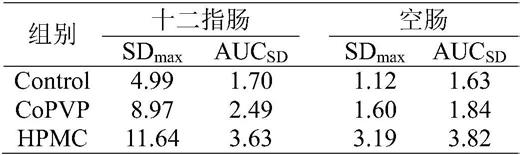
59.v
g
=v
s,0
×
e-(ln(2)
×
t/ge)
60.v
d
=v
d,0
61.v
j
=v
j,0
+v
s,0
x(1-e-(ln(2)
×
t/ge)
)+(v
g
+v
d
+v
j
)
×
t
62.此处v
g
、v
d
和v
j
分别代表胃腔室、十二指腔室和空肠腔室在时间t时的体积;v
(s,0)
、v
(d,0)
和v
(j,0)
分别表示胃腔室(300ml)、十二指腔室(50ml)和空肠腔室(100ml)生理相关介质的初始体积;ge表示胃半数排空期(8min);v
g
、v
d
和v
j
分别表示胃腔室、十二指腔室和空肠腔室分泌液泵入速度(均为1ml/min)。
63.阿帕替尼在模拟小鼠胃肠道的双相胃肠模拟系统中的吸收测试
64.溶液配制:
65.因为小鼠胃肠道与人体胃肠道ph值等有一定差异,而ph对bcs ii b类药物的超溶解与沉淀过程有较大影响,在建立体内外相关性时由此将产生较大的偏差。所以参考文献数据,将模拟十二指肠液、空肠液ph值分别调整为体内实验所用小鼠的生理值:4.7和5.0。
66.模拟十二指肠液(mouse fasted state simulated duodenal fluid,mfassdf):0.2mol/l磷酸氢二钠与0.1mol/l柠檬酸溶液两者以适当比例混匀调整ph为4.70。
67.模拟空肠液(mouse fasted state simulated jejunal fluid,mfassjf):0.2mol/l磷酸氢二钠与0.1mol/l柠檬酸溶液两者以适当比例混匀调整ph为5.00,加入2.240g fassif粉末溶解,静置2h即得。
68.模拟十二指肠分泌液(mouse fasted state simulated duodenal secretion,mfassds):按mfassdf方法以4倍浓度配制。
69.模拟空肠分泌液(mouse fasted state simulated jejunal secretion,mfassjs):按mfassjf方法以4倍浓度配制。
70.含量测定方法:
71.bgis在线监测:pion在线光纤紫外检测系统
72.二阶导range 335-340探头光程2mm 1mm分别建立标准曲线,
73.在相应不含药介质内扣除空白后,在线监测药物含量变化。
74.沉降抑制效能评价
75.图3中a为胃室,b为十二指肠室,c为空肠室,d为十二指肠吸收室,e为空肠吸收室,f药物吸收百分比f_vitro%;正方形代表仅有阿帕替尼组,三角形代表含copvp组,圆形代表含hpmc组,星形代表ph值,虚线代表当前ph值下对应色彩组别的饱和溶解度。(n=3)
76.药物体外吸收百分比f_
vitro
%=(c
do
×
v
do
+c
jo
×
v
jo
)/((c0×
v
(s,0)
))
×
100%
77.c
do
及c
jo
为十二指肠室、空肠吸收室药物浓度,v
do
及v
jo
为十二指肠室、空肠吸收相溶剂正辛醇体积。
78.图4a、图4b分别为十二指肠室和空肠室的过饱和度变化。
79.因为添加沉降抑制剂后,药物表观溶解度随之发生变化,因此对沉降抑制效果评价时,超溶解度(supersaturation degree)应当是所测浓度与对应组别溶解度之比值,即
80.sd=s
x,t
/s
x,0
81.可见hpmc组虽然浓度小于copvp组,但copvp因为是有提高阿帕替尼溶解度的作用,沉降抑制作用反而是hpmc更高,在十二指肠室中超溶解度最大值hpmc组sd
d,h,max
=9.82,copvp组sd
d,c,max
=6.98,阿帕替尼组sd
d,a,max
=3.98,空肠室内差异更为明显,超溶解度最大值hpmc组sd
j,h,max
=3.6,copvp组sd
j,c,max
=1.6,阿帕替尼组sd
j,a,max
=1.1。
82.sd
max
可以评价沉降抑制剂对超溶解的最大作用,为评价沉降抑制剂对超溶解的持续作用,采取指标超溶解度曲线下面积(area under curve of sd,auc
sd
)
83.评价结果见下表:
84.表1 copvp和hpmc沉降抑制作用效能指标评价结果
[0085][0086]
实施例3偏光显微镜与电镜观察
[0087]
按前述方法配制阿帕替尼及各组溶液,与模拟肠液按1∶2混匀后取1滴于带凹槽载玻片上,盖玻片小心覆盖液滴,于同一视野下连续观察沉淀过程。
[0088]
取各体外沉降模拟实验所得沉淀物,3800r/min离心10min,30℃真空干燥过夜。取适量沉淀物于铜板上,供扫描电镜拍摄沉淀物形态。
[0089]
设备信息:日本电子公司,扫描电子显微镜,型号:jsm-6510。
[0090]
实验操作过程:取适量干燥样品粘至样品台上,通过离子溅射仪在样品表面镀一层20nm左右的金膜。然后将样品移至扫描电镜样品仓中抽真空,当真空达到测试要求后,加高压发射电子,聚焦电子束与样品相互作用,激发二次电子发射,二次电子发射量随样品表面形貌变化而变化,通过探测器采集二次电子得到反映样品表面形貌的二次电子像。加速
电压选择10kv或15kv。
[0091]
图5为阿帕替尼沉淀过程显微图。a阿帕替尼组,b含copvp组,c含hpmc组,1、2、3分别为5min、60min、120min。结果表明阿帕替尼可以快速形成晶体,而copvp和hpmc能明显抑制晶体生成。
[0092]
图6为阿帕替尼沉淀物电镜图。a为阿帕替尼组;b为含copvp组;c为含hpmc组。可见阿帕替尼沉淀形成簇晶,copvp因高分子网状结构隔绝沉淀以细小微粒避免形成较大晶体,hpmc具有一定抑制作用,沉淀颗粒为较小方晶。各组结果与偏光显微镜结果对应。
[0093]
实施例4 xrd和dsc分析
[0094]
取各体外沉降模拟实验所得沉淀物,3800r/min离心10min,30℃真空干燥过夜。取适量沉淀物于单晶硅板,供xrd分析。
[0095]
取约2mg沉淀物于5mm铝制坩埚,精密称重后封盖供dsc分析。
[0096]
xrpd测试条件:bruker d8 discovery xrpd(40kv,40ma,2-theta mode,start10
°
,increment19
°
,end48
°
)
[0097]
dsc测试条件:mettler toledo dsc 3+(25℃-300℃,10℃/min,n2 50ml/min purge gas)
[0098]
图7显示了阿帕替尼api、甲磺酸阿帕替尼和通过溶剂转移实验收集的沉淀物样品的dsc热谱图,其中不使用沉降抑制剂(pi)(a)、使用copvp(b)、使用hpmc(c)、甲磺酸阿帕替尼(d)和阿帕替尼碱(e)吸热峰分别为153.15℃、152.70℃、152.70℃、201.62℃和167.99℃。沉淀物的熔点比甲磺酸阿帕替尼粉末低约50℃,甚至比普通阿帕替尼碱晶型低15℃。
[0099]
图8为阿帕替尼沉淀的x射线衍射图,无pi(a),copvp(b),hpmc(c),nacl(d),阿帕替尼游离碱(e),甲磺酸阿帕替尼(f),copvp(g)和hpmc(h)。
[0100]
实施例5阿帕替尼在小鼠体内吸收测试
[0101]
实验动物:
[0102]
c57bl/6雄性小鼠购于常州卡文斯实验动物有限公司,许可证号scxk(苏)2016-0010,spf级。
[0103]
配制溶剂:
[0104]
称取阿帕替尼10.2mg溶于丙二醇8ml,再加入生理盐水12ml,混匀,即得含阿帕替尼0.5mg/ml供注射的a液。称取阿帕替尼50.6mg溶于50ml ph 2hcl溶液酸液中,即得含阿帕替尼1mg/ml供灌胃的b液;取b液20ml溶copvp 200mg,即得含1%copvp和阿帕替尼1mg/ml的灌胃c液;取b液20ml溶hpmc 200mg,即得含1%hpmc和阿帕替尼1mg/ml的灌胃d液。
[0105]
c57bl/6小鼠雄性,体重20
±
2g,禁食不禁水一夜后,随机分为4组,静注组以尾静脉注射a液5ml/kg(即2.5mpk);其余灌胃10ml/kg(即10mpk)分别为不含沉降抑制剂组,含copvp组,含hpmc组。给药后0.083、0.25、0.5、1、2、4、6h,眼眶取血约50μl,于edta-k2化ep管中,4000rpm离心10min后取上清测定。
[0106]
血药含量测定:
[0107]
仪器
[0108]
高效液相色谱-质谱联用系统:shimadzu cbm30alite液相系统(含shimadzu lc30ad泵,dgu-20a3在线真空脱气机,shimadzu sil30ac恒温自动进样器,shimadzu cto20ac柱温箱);sciex triple quad
tm
qtrap 5500mass spectrometer系统(esi接口离子
源);g560e型旋涡混合器(美国scientific industries,inc),legend micro 21r型冷冻高速离心机(thermo fishier公司);梅特勒托利多xse204电子天平;milli-qa10超纯水器(美国millipore公司)。
[0109]
供试品制备
[0110]
对照品溶液:精密称取阿帕替尼10.13mg,溶于甲醇10ml,配成1mg/ml储备液,用50%甲醇-水稀释成浓度为10、30、100、300、1000、3000ng/ml的系列标准液。
[0111]
质控溶液:精密称取阿帕替尼10.15mg,溶于甲醇10ml,配成1mg/ml储备液,用50%甲醇-水稀释成浓度为20、200、2400ng的系列质控溶液。
[0112]
内标溶液:精密称取吉非替尼10.18mg,溶于10ml甲醇,配成1mg/ml储备液,用50%甲醇-水稀释成浓度10ng的内标液。
[0113]
标准曲线溶液:依次加入50μl内标液、10μl空白血浆、10μl各对照品溶液,涡旋混匀,再加入430μl乙腈,涡旋混匀、10000rpm离心10min,取适量上清液置于进样瓶中内插管,备用。
[0114]
将对照品溶液替换为质控样品,得到3个浓度的质控样品;将标准溶液替换为50%甲醇-水,得到blank液;将内标液、对照品溶液替换为50%甲醇-水,double blank液;将空白血浆替换为各待测血浆、对照品溶液替换为50%甲醇-水,得到各供试品。
[0115]
色谱条件
[0116]
色谱柱hypersil gold(50mm
×
2.1mm,3μm),5mmol/l乙酸铵水溶液:乙腈=40:60(v/v),流速0.4ml/min,柱温40℃,进样量5μl。
[0117]
质谱条件
[0118]
离子源为电喷雾电离源(esi),正离子模式检测,毛细管电压,干燥气温度,干燥气流速,雾化气压力选择性监测质荷比(m/z)q1 mass(da):398.3,q3 mass(da)184.3(为阿帕替尼)和q1 mass(da):447.1,q3 mass(da)128.1(内标物吉非替尼)的离子峰,碎片电压ce(volts)分别为32和50,time:100msec。
[0119]
图9为阿帕替尼各组药时曲线(附图各组生物利用度),
■
表示无pi组(blank),
▲
表示含copvp组,
●
表示含hpmc组,
◆
表示阿帕替尼(i.v.)组。
[0120]
表3.阿帕替尼的药代动力学参数
[0121][0122]
结果表明copvp、hpmc能显著提高阿帕替尼生物利用度,阿帕替尼的生物利用度从39%增至60%和52%,即相对生物利用度提高151%和133%。也能明显抑制晶体生成。(*p<0.05,***p<0.001)
[0123]
实施例6ivivc构建
[0124]
基于gastroplus软件通过静注药时曲线得到pk参数,将其代入口服药时曲线并去卷积,则得到口服药物在体内吸收分数-时间曲线fa%-t;将药物体外吸收百分比-时间曲m%-t与fa%-t作levy plot,得到缩放参数,据此对m%-t进行缩放;将缩放后的m%-t进行
卷积操作,得到预测药时曲线(c-t)。
[0125]
图10为两种有机相的总和随时间的体外溶解度:正方形
■
表示无pi,三角形
▲
表示copvp,圆圈
●
表示hpmc;实线是双威布尔模型的拟合曲线。
[0126]
图11为通过loo-riegelman反卷积随时间吸收的馏分。
[0127]
图12为levy图:正方形
■
代表没有pi,三角形
▲
代表copvp,虚线是回归。
[0128]
图13为体外溶解的分数与体内吸收的分数之间的相关性,正方形
■
表示无pi,三角形
▲
表示copvp,虚线是回归。
[0129]
图14为预测和实际药物血浆浓度-时间曲线:正方形
■
表示无pi,三角形
▲
表示copvp,圆圈
●
表示hpmc。具有三种颜色的实线代表每组中阿帕替尼的相应预测血浆浓度。
[0130]
表4.基于相关模型的c max和auc值的预测误差
[0131][0132]
从表4可知,内部和外部验证的pe均小于10%,达到了a级ivivc标准。结果表明,bgis是体外评估阿帕替尼过饱和度同时筛选pi的合适装置。通过体外实验预测体内性能,该ivivc可用于建立评估沉淀抑制剂对阿帕替尼过饱和的作用的规范。选择合适的pi甚至筛选合适的配方将节省成本和时间。由于内部积分方法差异,由gastroplus获得的观察到的auc比由winnonlin获得的平均auc略高。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1