基于深度学习的抗原亲和力预测方法和系统与流程
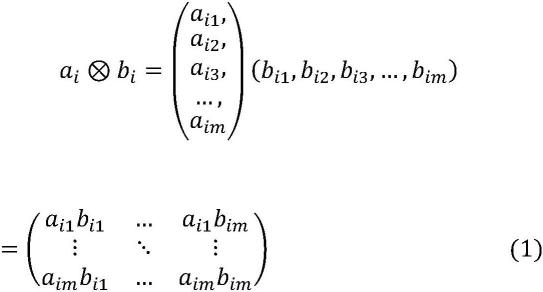
1.本发明属于人工智能领域,更具体而言本发明涉及一种基于深度学习的抗原亲和力预测方法和系统。
背景技术:
2.抗原表位(antigenic determinant,ad),指抗原分子中决定抗原特异性的特殊化学基团。抗原通过抗原表位与相应的淋巴细胞表面的抗原受体结合,从而激活淋巴细胞,引起免疫应答。抗原表位的性质、数目和空间构型决定了抗原的特异性。抗原表位的大小与相应抗体的抗原结合部位相适合。一般情况下,一个多肽表位含5~6个氨基酸残基;一个多糖表位含5~7个单糖;一个核酸半抗原的表位含6~8个核苷酸。一个抗原表位的特异性由组成它的所有残基共同决定,但其中有些残基在与抗体结合时比其它残基起更大作用,这些残基被称为免疫显性基团。
3.t细胞表位为免疫原性多肽片段,必须经抗原呈递细胞加工处理为小肽分子,然后再与主要组织相容性复合体(major histocompatibility complex,mhc)分子结合才能被t细胞所识别。t细胞抗原受体(t cell receptor,tcr)仅能识别约10~20个氨基酸左右的小分子多肽。这些表位由序列上相连的氨基酸组成,主要存在于抗原分子的疏水区,称为线性表位或序列表位。由于t细胞仅能识别经加工处理的表位,故一般不识别天然抗原的构象型表位。
4.抗原表位的分析方法多种多样,包括化学裂解法、生物酶解法、核磁共振光谱(nmr)、表面等离子共振技术(spr)、杂交肽裂解法、多肽文库构建法、理论测法等等。目前,随着计算机技术的发展,尤其是人工智能技术的普及,从生物学大数据中直接对抗原表位进行筛选预测成为了通量最高、成本最低、周期最短的抗原表位分析方法。
5.就t细胞表位而言,理论预测的常规方案是通过对mhc分子与抗原肽结合数据的学习,提取众多关键特征,如抗原肽序列、抗原肽长度、抗原肽表达量、抗原肽热稳定性等,从而形成机器学习模型,结合高通量测序技术,对未知蛋白或基因的潜在抗原肽进行理论预测。目前国内外已有多家公司相继开发了抗原预测软件,如丹麦科技大学开发的netmhcpan软件、gritstone oncology公司开发的edge软件、武汉华大吉诺因生物科技有限公司开发的epip软件等。
6.随着现代生物信息学、分子生物学和分子免疫学的不断发展和相互促进,表位研究及其应用已经取得了长足进步,并展现出巨大的应用潜力。抗原表位的应用主要体现在三个方面,即疾病诊断、疫苗开发和疾病治疗。
7.在疾病诊断中,表位诊断方法效率的关键是敏感性与特异性。抗原表位是激发免疫反应的基本单元,单一表位激发特异性单一免疫反应,而多重表位往往激发多重混合免疫反应,进而产生非特异性混合抗体、致敏淋巴细胞或效应因子。因而,疾病诊断中的表位肽研究注重于特异性表位肽的选择,从而达到更好的诊断效能。
8.在疫苗开发中,常规疫苗均包含大量抗原表位,这些表位包括保护性表位、抑制性
表位或无效表位。只有在保护性表位诱导的免疫反应占优势地位时,疫苗才能达到理想的保护性效果。因而,在既往抗病毒疫苗的研究中,如何获取免疫原性强、序列保守性强、对病毒入侵起关键作用的蛋白表位往往是技术难点。
9.在疾病治疗中,表位诱导的免疫反应具有高度特异性与针对性,可用于肿瘤、感染性疾病和自身免疫性疾病的免疫治疗。免疫治疗通过增强患者自身免疫系统来激活对抗原的细胞毒性反应,在近年来已被证实为一种有效的策略。该策略利用了来自细胞内蛋白酶体裂解的各种突变蛋白形成的细胞表面多种抗原。这些多肽与hla分子结合,形成多肽-hla复合物呈递给t细胞受体(tcr)。如果tcr能识别这些多肽-hla复合物,则会激活细胞毒性t淋巴细胞(cytotoxic lymphocyte,ctl),该细胞是白细胞的亚部,为一种特异t细胞,专门分泌各种细胞因子参与免疫作用,对某些病毒、肿瘤细胞等抗原物质具有杀伤作用,与自然杀伤细胞构成机体抗病毒、抗肿瘤免疫的重要防线。细胞毒性t淋巴细胞治疗的第一步是预测抗原与hla分子的结合亲和力。目前肿瘤治疗领域中进展迅速的新抗原(neoantigen)疗法就是表位在疾病治疗领域的良好应用。国外已有多家公司开展新抗原治疗恶性肿瘤研究,并进入临床实验阶段,如biontech、neon therapeutics、gritstone oncology等。
10.目前已有四种预测抗原与hla分子亲和力的方法,分别是基于结构的方法、基于机器学习的方法、基于位置权重矩阵(pssm)的方法和组合方法。其中基于机器学习的方法通过已知的结合肽和非结合肽信息学习高维分类平面来预测多肽结合的亲和力。机器学习的方法能够准确预测多肽与特异性hla等位基因的亲和力,比如hla-a*0201、hla-a*0101和hla-b*0702[1,2],因此在许多研究中经常被使用[3-5]。其中最主流的机器学习算法是泛特异性亲和力算法,该算法同时将hla的分子氨基酸序列和多肽的氨基酸序列作为输入,通过机器学习算法得到亲和力预测输出,目前业界最常用的算法是netmhcpan[6],该方法通过一段34位氨基酸伪序列(pseudo sequence)来表征hla分子,然后预处理该伪序列与多肽短序列(peptide sequence),并列预处理结果作为输入特征,再通过bp神经网络模型(back propagation,bp)得到多肽-hla分子亲和力预测,这种方法将每个多肽-hla分子对建模为独一无二的输入序列,从而可以通过模型学习该序列和多肽-hla分子亲和力值的映射关系,这种做法业界称为泛特异化训练策略(pan model)。netmhcpan工作显示单一bp模型效果不佳,为了得到最好的效果,netmhcpan通常会同时聚合学习大量模型。
[0011]
近年来业界出现了许多基于深度学习的亲和力预测模型,其中比较有代表性的算法是deepseqpan[7],该方法通过两套独立的卷积网络分别学习多肽和hla特征,合并特征之后通过神经网络预测亲和力输出;类似的思路被用于mhcseqnet[8]和ai-mhc[9],mhcseqnet使用门控循环单元(gated recurrent units,gru)代替卷积神经网络,目的是学习可变长度的多种多肽数据,ai-mhc通过填充(padding)来处理不同的多肽,从而使用效率更高的卷积神经网络实现变长输入学习;类似的工作还有acme[10],与deepseqpan的区别在于拼接每个hla计算层都会拼接多肽表征向量;除了使用自然语言处理技术,convmhc[11]使用了一种类图像处理的思路建模亲和力模型:将多肽和hla分子的位置理化数据建立为多个2维的数据矩阵,采用2维卷积神经网络预测亲和力模型。
[0012]
现有技术的主要不足之处在于:1)缺乏有效的方式建模表达多肽-hla复合物。上述方法中,[6-10]通过独立的神经网络学习多肽和hla分子的序列特征,然后直接拼接为一体,模型设计时并没有考虑多肽和分型一旦呈递会产生的复合物结构;2)多肽序列较短,直
接使用深层网络效果较差,相关工作大多采用浅层网络。[6]采用大量传统神经网络聚合弥补单模型学习力弱的缺陷;[7,9,10]都使用了浅层卷积网络用于学习多肽特征;3)某些方法没有考虑多肽长度多样性。[11]虽然能将深层网络用于多肽学习,但是设计上只能考虑定长,不能做到可变长度;同样的问题出现在[6]中。
[0013]
因此,本发明需要改进的基于深度学习的亲和力预测模型。
技术实现要素:
[0014]
基于现有技术中存在的问题,本发明的目的在于提供一种改进的基于深度学习的亲和力预测模型及其构建方法。
[0015]
因此,在第一方面中,本发明提供了一种基于深度学习抗原亲和力的方法,所述方法包括:
[0016]
(1)获取低聚体-免疫分子的结合结构的训练数据集,所述训练数据集包括低聚体-免疫分子结合的亲和力;
[0017]
(2)对于待测低聚体与免疫分子和所述低聚体-免疫分子的结合结构的训练数据集中的每一组,将低聚体的序列、低聚体的单体位置和免疫分子分别表示为高维向量,并将所述三个高维向量融合成低聚体-免疫分子的结合结构的向量;
[0018]
(3)利用所述训练数据集中低聚体-免疫分子的结合结构的向量和亲和力数据训练深度神经网络,建立低聚体-免疫分子亲和力预测模型;
[0019]
(4)将所述待测低聚体-免疫分子的结合结构的向量输入所述抗原亲和力预测模型,利用训练的深度神经网络预测所述待测低聚体-免疫分子结合的亲和力。
[0020]
在一个实施方案中,所述低聚体选自:多肽、多糖和核酸半抗原。
[0021]
在一个实施方案中,所述低聚体为变长长度的向量。
[0022]
在一个实施方案中,所述免疫分子为主要组织相容性复合体分子,例如人类白细胞抗原,包括但不限于i类和ii类人类白细胞抗原。
[0023]
在一个实施方案中,所述低聚体-免疫分子结合的亲和力表示为低聚体-免疫分子结合的ic
50
值。
[0024]
在一个实施方案中,在(2)中,通过词嵌入获得能够反映任意两个单体之间关系的单体向量表示。
[0025]
在一个实施方案中,在(2)中输入低聚体序列为(x1,x2,
…
,xn),其中xi为氨基酸字符,xi∈{a,c,d,...,y},映射结果是将xi转变为一个m维向量zi∈rm,r为实数空间,序列(x1,x2,...,xn)转变为(z1,z2,...,zn)。
[0026]
在一个实施方案中,在(2)中单体位置表征向量:将多肽每个氨基酸位置的标记,映射到高维连续数值空间:输入统一为向量(1,2,...,n),每个位置i被全连接神经网络映射成一个向量pi∈rm,r为实数空间,即(1,2,...,n)输出为(p1,p2,...,pn)。
[0027]
在一个实施方案中,在(2)中所述低聚体序列和所述低聚体的单体位置的高维向量相加形成所述低聚体的表征向量。
[0028]
在一个实施方案中,在(2)中免疫分子向量的输入为(y1,y2,...,yk),yi∈{a,c,d,...,y},输出为(z
′1,z
′2,...,z
′n),z
′i∈rm,从k维原始氨基酸向量映射为与低聚体格式相同的n
×
m维向量。
[0029]
在一个实施方案中,在(2)中将所述低聚体的表征向量和所述免疫分子的向量进行张量乘法、张量加法或注意力运算。
[0030]
在一个实施方案中,在(3)中所述深度神经网络包括深层卷积层和多层全连接层,所述深度卷积层用于提取训练数据集中低聚体-免疫分子的特征向量,输入所述多层全连接层,所述多层全连接层将所述特征向量映射为亲和力值,通过反向传播算法得到网络参数。
[0031]
在一个实施方案中,对于每个深层卷积层,将输入复制3份,两份通过不同的卷积网络学习,并且对其中一个卷积输出做sigmoid归一化操作,从而得到两个向量a和b,按位乘法得到残差a
×
b;另一份与a
×
b的残差做加法。
[0032]
在一个实施方案中,在最后一层卷积层之后添加一个全局池化层θ:rn×
p
→rp
,p为最后一层的输出维度,得到特征向量。
[0033]
在一个实施方案中,在所述多层全连接层中,通过全连接层将深层卷积层得到的特征向量映射为亲和力值,方法如下:
[0034]
1)将深层卷积层的输出特征向量x做线性变换,作为全连接层的输入向量y=wx+b;
[0035]
2)使用线性整流函数对线性变换结果做非线性转换:
[0036][0037]
1)和2)构成一层映射网络,经过多层映射网络输出亲和力预测结果。
[0038]
在一个实施方案中,在(4)中,所述训练的深度神经网络包括深层卷积层和多层全连接层,所述深度卷积层用于提取所述待测低聚体-免疫分子的特征向量,输入所述多层全连接层,预测所述待测低聚体-免疫分子结合的亲和力。
[0039]
在第二方面中,本发明提供了一种基于深度学习建立抗原亲和力预测模型的方法,所述方法包括通过本发明第一方面的方法的(1)-(3)建立低聚体-免疫分子亲和力预测模型。
[0040]
在第三方面中,本发明提供了利用本发明第二方面的方法建立的抗原亲和力预测模型,所述抗原亲和力预测模型包括:数据获取模块、输入向量建立模块和模型建立模块,
[0041]
所述数据获取模块,用于获取低聚体-免疫分子的结合结构的数据集,所述数据集包括低聚体-免疫分子结合的亲和力;
[0042]
所述输入向量建立模块,用于利用所述低聚体-免疫分子的结合结构的数据集,将所述低聚体的序列、所述低聚体的单体位置和所述免疫分子分别映射为高维向量,并将所述三个高维向量融合成所述低聚体-免疫分子的结合结构的向量;
[0043]
所述模型建立模块,用于利用所述低聚体-免疫分子的结合结构的向量和亲和力数据训练深度神经网络,建立抗原亲和力预测模型。
[0044]
在一个实施方案中,在模型建立模块中,所述深度神经网络包括深层卷积层和多层全连接层,所述深度卷积层用于提取训练数据集中低聚体-免疫分子的特征向量,输入所述多层全连接层,所述多层全连接层将所述特征向量映射为亲和力值,通过反向传播算法得到网络参数。
[0045]
在一个实施方案中,对于每个深层卷积层,将输入复制3份,两份通过不同的卷积
免疫分子的结合结构的向量;(3)利用所述训练数据集中低聚体-免疫分子的结合结构的向量和亲和力数据训练深度神经网络,建立低聚体-免疫分子亲和力预测模型。
[0060]
优选地,所述深度神经网络包括深层卷积层和多层全连接层,所述深度卷积层用于提取训练数据集中低聚体-免疫分子的特征向量,输入所述多层全连接层,所述多层全连接层将所述特征向量映射为亲和力值,通过反向传播算法得到网络参数。
[0061]
在一个实例中,在多层卷积层中,每层卷积神经网络学习上一层残差:第i层输入记为x,第i层卷积网络为convi,则第i层输出为x+convi(x);即将输入复制3份,两份通过不同的卷积网络学习,并且对其中一个卷积输出做sigmoid归一化操作,从而得到两个向量a和b,按位乘法得到残差(a
×
b);另一份与a
×
b的残差做加法。
[0062]
在一个实例中,在多层全连接层中,通过全连接网络将计算网络学习到的特征向量映射为亲和力值,方法如下:
[0063]
1)将映射网络的输入(即计算网络的输出)做线性变换,将映射层的输入向量x转化为y=wx+b;
[0064]
2)使用线性整流函数(relu)对线性变换结果做非线性转换:
[0065][0066]
1)和2)构成一层映射网络,经过多层输出亲和力预测结果。
[0067]
以下通过多肽-hla分子的建模和应用对本发明进行示例性说明。本发明中使用的多肽-hla分子数据集来自iedb公开数据。
[0068]
在本发明中,泛特异性模型(pan model)需要将多肽和hla分子序列文本映射为一个独一无二的向量,从而让神经网络模型建立向量与预测指标之间的函数映射关系;除此之外,映射过程需要建模多肽与hla分子之间的结合结构。
[0069]
本发明的深度神经网络模型主体由三部分组成,参考图1:由输入网络、计算网络和映射网络三部分组成一个神经网络整体。在模型训练过程中,首先需要通过训练过程得到网络的参数,训练过程使用已知亲和力的低聚体-免疫分子的结合结构的向量数据,通过反向传播算法得到网络参数;在测试/预测过程,将待预测的低聚体-免疫分子的结合结构的向量输入至输入网络,经计算网络、映射网络输出预测的亲和力值。下面是具体功能介绍。
[0070]
输入网络:
[0071]
输入网络的功能是将多肽和hla分子通过神经网络建立一个高维向量,建模多肽-hla分子结合结构,实现模型的泛特异性输入。不同的多肽+hla分子被映射为不同的向量,高维度目的是充分捕捉不同输入序列的细微区别。这里发明人输入的是hla分子伪序列,这样准确率更高。
[0072]
具体设计参考图2。多肽序列和hla分子序列的每一位氨基酸分子被全连接网络映射成更高维向量a;将多肽位置标记为1~多肽长度,对位置序列通过另一个网络映射到同样的维度;将向量扩张成图中的张量然后进行张量运算,得到混合了多肽和hla分子信息的最终输入张量。图2中举例为15多肽长度,128维映射输出维度。图2中上4下3的全连接圈表示的是“每个氨基酸分子从a~z的字符空间映射到高维连续数值空间”,这是一个常用的全连接层表示法,上面的圈代表了全连接输入维度即氨基酸字符空间,下面的圈代表了全连
接输出维度即更高维向量。是以15长度的多肽为例,每个位置从氨基酸字符映射成连续的高维向量,这里的高维维度也举例为m。15
×
128
×
1是将向量扩张为张量的过程,目的是完成图中的张量加法和张量乘法。张量运算的原则是以后两个维度为矩阵做运算,前面的维度看作批大小(batch size),15
×
128
×
1的两个张量加法后仍然是15
×
128
×
1,与15
×1×
128的张量做乘法后结果为15
×
128
×
128。
[0073]
可以利用语言模型[12-15]对于文本序列的词嵌入(word embedding)工作,将原始输入数据通过神经网络映射为三个高维向量。这里以多肽长度n,hla分子长度k,输出维度映射为m维为例:
[0074]
1)多肽表征向量通过全连接神经网络,将多肽原始序列作为一个变长的向量,向量的每个氨基酸分子从a~z的字符空间映射到高维连续数值空间。输入多肽序列为(x1,x2,...,xn),其中xi为氨基酸字符,xi∈{a,c,d,...,y},映射结果是将xi转变为一个m维向量zi∈rm,m是一个需要交叉验证调试的参数,r为实数空间,序列(x1,x2,...,xn)转变为(z1,z2,...,zn);
[0075]
2)肽位表征向量:通过全连接神经网络,将多肽每个氨基酸位置的标记,映射到高维(例如m维)连续数值空间:输入统一为向量(1,2,...,n),每个位置i被全连接神经网络映射成一个向量pi∈rm,r为实数空间,即(1,2,...,n)输出为(p1,p2,,..,pn);
[0076]
3)hla分子表征向量:每条多肽对应的hla分子也需要被映射到高维连续数值空间,考虑到hla分子和多肽每一位都是氨基酸分子,具有相同意义,这里映射使用的神经网络模型与多肽表征向量相同。hla分子向量的输入为(y1,y2,...,yk),yi∈{a,c,d,...,y},输出为(z
′1,z
′2,...,z
′n),z
′i∈rm,从k维原始氨基酸向量映射为与多肽格式相同的n
×
m维向量,过程分为如下几步:
[0077]
a)通过1)的神经网络将(y1,y2,...,yk)映射为(y
′1,y
′2,...,y
′k),y
′j∈rm;
[0078]
b)对(y
′1,y
′2,...,y
′k)取平均得到复制n份得到(z
′1,z
′2,...,z
′n),),
[0079]
在融合上述三种向量时可以使用两种机制,一种是类似传统语言模型对三个向量做加法。然而,基因组序列不同于语言,多肽和hla分子具有空间上的结合结构,需要用更复杂的机制捕捉,因此还可以使用另一种机制:
[0080]
1)计算多肽的表征向量a=(z1+p1,z2+p2,...,zn+pn);
[0081]
2)对a的每个元素ai,执行张量扩张(克罗内克积):rm→rm
×1;
[0082]
3)将hla分子表征向量,记为b,对b的每个元素bi,执行张量扩张(克罗内克积):rm→
r1×m;
[0083]
4)对a、b做按位张量积其中:
[0084][0085]
5)对做打平(flatten)操作。即对于n位中每一位元素将式(1)结果转化为m2维向量(a
i1bi1
,...,a
i1bim
,...,a
imbi1
,...,a
imbim
)。该向量为计算网络最终输入。
[0086]
通过神经网络将多肽、hla分子映射到同一个高维空间,通过张量乘积生成多肽和hla分子序列的立体结构映射,为高效率泛特异性亲和力模型提供输入。
[0087]
对于训练数据而言,需要将计算网络与映射网络结合作为一个整体完成学习过程。通过计算网络的深层卷积神经网络和映射网络的全连接网络,将低聚体-免疫分子的结合结构的向量和亲和力值输入计算网络和映射网络进行训练进行,完成计算网络和映射网络参数的学习。
[0088]
计算网络:
[0089]
计算网络包括多层卷积神经网络,其功能是通过深层卷积神经网络有效提取短序列特征,得特征向量。因为输入的多肽和hla分子长度很短(i型hla对应多肽通常小于15位,ii型hla通常小于26位),低分辨率数据会导致深层卷积神经网络的过拟合,使得深层卷积神经网络的效果并不佳。为了加大深层卷积神经网络复杂度以强化学习能力,提高使用深层卷积神经网络的效果,使用残差神经网络结合门控线性单元(gated linear unit)激活(参见[15]),显著增强深层卷积神经网络(例如至少5层及以上,优选10层及以上,例如15层以上,甚至20层以上)的泛化能力,提升复杂度,强化算法学习能力,具体设计参考图3。将输入复制3份,两份通过不同的卷积网络学习,并且对其中一个卷积输出做sigmoid归一化操作,从而得到两个向量a和b,按位乘法得到残差(图中a
×
b);另一份与a
×
b的残差做加法。通过采用上述残差网络设计(图3),每层卷积神经网络学习上一层残差:第i层输入记为x,第i层卷积网络为convi,则第i层输出为x+convi(x)。通过学习残差而不是学习输入本身的策略,有效降低了卷积神经网络加层带来的过拟合效应,最终的计算网络包括例如10层卷积神经网络,同时使用门控线性单元作为激活机制,整体流程如下:
[0090]
1)对于输入x=(x1,x2,...,xn),分别输入两个卷积神经网络conva和convb,两个卷积神经网络的输出维度与输入x一致;
[0091]
2)分别计算得到两个卷积神经网络输出conva(x)和convb(x),对其中一个卷积神经网络的输出按位做sigmoid映射σ:r
→
[0,1],例如对于convb:
[0092][0093]
3)按位乘conva(x)i和σ(convb(x)i)得到(conva(x)1×
σ(convb(x)1),conva(x)2×
σ(convb(x)2),...,conva(x)n×
σ(convb(x)n)),与x=(x1,x2,...,xn)做向量加法,得到计算层输出,通过dropout层后传递至下一层;
[0094]
考虑到实际中多肽长度是可变的,因此通过所有卷积层之后的输出长度也是可变的,在最后一层卷积神经网络之后会添加一个全局池化层θ:rn×
p
→rp
,p为最后一层的输出维度,得到特征向量。
[0095]
针对氨基酸序列的深度卷积神经网络模型,通过学习每层卷积神经网络输出的残差而不是输出本体的设计,提升深层卷积残差网络结构的泛化性,使得单个模型就能达到良好准确率,从而显著提升整体流程效率。
[0096]
映射网络:
[0097]
映射网络包括多层,其功能是通过全连接网络将计算网络学习到的特征向量映射为亲和力值值,整体流程如下:
[0098]
1)将映射网络的输入(即计算网络的输出的特征向量)做线性变换,将映射网络的输入向量x转化为y=wx+b;
[0099]
2)使用线性整流函数(relu)对线性变换结果做非线性转换:
[0100][0101]
1)和2)构成一层映射网络,经过多层映射网络输出亲和力预测结果。
[0102]
对于测试/预测数据而言,将待测试/预测的多肽+hla分子输入所述训练的计算网络的深层卷积神经网络,根据网络的参数,提取低聚体-免疫分子的特征向量。输入所述映射网络,所述映射网络的参数,所述映射网络的多层全连接层将所述特征向量映射为亲和力值。在一个实例中,亲和力值可以为ic
50
。
[0103]
以下通过实施例验证本发明的抗原亲和力预测模型的效果。
[0104]
实施例
[0105]
算法使用无需显卡资源,但是需要配置相应的软件环境:
[0106]
python:3.6.5
[0107]
anaconda:4.5.4
[0108]
tensorflow:1.3.0
[0109]
cuda:8.0
[0110]
cudnn:6.0
[0111]
发明人在使用时,通过执行本软件提供的预测脚本:python predict.py待预测文件路径,就会生成每条实验数据的预测结果。待预测的文件格式需要整理为
″
mhc,peptide
″
格式。发明人在代码中放了一个实例文件,在data文件夹下的test_batch.csv,如下:
[0112][0113]
该测试数据是iedb数据库mhc ligand assay方法验证的pmhc(9、10mer)亲和力定量数据集(http://www.iedb.org/),分型包括a0101、a0201、a2402等共113种hla分型。使用该脚本会在result文件夹下生成predict.csv文件。该文件记录了每条样本的多肽、分型类
dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specifc t cells.science 2015;348l803-8.
[0124]
[4]walter s,weinschenk t,stenzl a,et al.multipeptide immune response to cancer vaccine ima901 after single-dose cyclophosphamide associates with longer patient survival.nat med 2012;18:1254-61.
[0125]
[5]yadav m,jhunjhunwala s,phung qt,et al.predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing.nature 2014;515:572-76.
[0126]
[6]nielsen,m.,andreatta,m.netmhcpan-3.0;improved prediction of binding to mhc class i molecules integrating information from multiple receptor and peptide length datasets.genome med 8,33(2016)doi:10.1186/s13073-016-0288-x.
[0127]
[7]liu,z.,cui,y.,xiong,z.et al.deepseqpan,a novel deep convolutional neural network model for pan-specific class i hla-peptide binding affinity prediction.sci rep 9,794(2019)doi:10.1038/s41598-018-37214-1.
[0128]
[8]phloyphisut,p.,pomputtapong,n.,sriswasdi,s.et al.mhcseqnet:a deep neural network model for universal mhc binding prediction.bmc bioinformatics 20,270(2019)doi:10.1186/s12859-019-2892-4.
[0129]
[9]sidhom,j.-w.a.p.d.a.b.a.ai-mhc:an allele-integrated deep learning framework for improving class i&class ii hla-binding predictions.biorxiv,p.318881(2018).
[0130]
[10]yan hu,ziqiang wang,hailin hu,fangping wan,lin chen,yuanpeng xiong,xiaoxia wang,dan zhao,weiren huang,jianyang zeng,acme:pan-specific peptide-mhc class i binding prediction through attention-based deep neural networks,bioinformatics.
[0131]
[11]han,youngmahn&kim,dongsup.(2017).deep convolutional neural networks for pan-specific peptide-mhc class i binding prediction.bmc bioinformatics.18.585.10.1186/s12859-017-1997-x.
[0132]
[12]devlin j,chang m w,lee k,et al.bert:pre-training of deep bidirectional transformers for language understanding[j].arxiv preprint arxiv:1810.04805,2018.
[0133]
[13]vaswani a,shazeer n,parmar n,et al.attention is all you need[c]//advances in neuralinformation processing systems.2017:5998-6008.
[0134]
[14]gehring j,auli m,grangier d,et al.convolutional sequence to sequence learning[c]//proceedings of the 34th international conference on machine learning-volume 70.jmlr.org,2017:1243-1252.
[0135]
[15]dauphin y n,fan a,auli m,et al.language modeling with gated convolutional networks[c]//proceedings of the 34th international conference on machine leaming-volume 70.jmlr.org,2017:933-941.
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1