一种当归川芎组合物及其制备方法与流程
本发明属中药技术领域,具体而言,本发明涉及一种当归川芎组合物及其制备方法。
背景技术:
佛手散出自宋许叔微《普济本事方》,是古代妇产科治疗胎前、产后诸症的要方。如神通广大之佛手,故名“佛手散”。其原文记载“当归(六两,洗)、川芎(四两,洗)上粗末,每服二钱,水一小盏,煎令泣泣欲干,投酒一大盏,止一沸,去滓温服”。“能治妊孕五七月,因事筑磕著胎,或子死腹中,恶露下,流血不止”。
该方剂中仅由当归和川芎两味中药组成,二者配伍,既是古方,又是著名对药,古今应用皆以其“养血活血,去淤生新“为其作用基础。该方剂现已被广泛应用于与之辩证相关的病症,如原发性及青春期痛经的治疗、引产下胎、产后淤血、流血腹痛等妇科疾病,偏头痛及顽固性疼痛头痛等脑神经病,心脑血管病及肝胆疾病等领域,具有深远的临床开发和应用价值。
现代植化分离及药理研究对其各提取部位及活性成分进行了系统研究,其主要活性物质为苯酞类、有机酚酸类、生物碱类、黄酮类,及多糖类化合物,按提取部位主要集中在挥发油、醇提液、水提液、水提醇沉液等部位。其中挥发油类成分含量高且药效活性强,是主要的药效物质,但容易挥发和降解,稳定性差;而采用水、醇系统提取中、高极性成分时,常伴随鞣质、色素、树胶等无效成分的一并提出,浓缩时,造成浸膏粘稠、出膏率高的现象,进而影响制剂的可行性和药效的发挥。这些都是本方发展为现代中成药制剂所要面临的技术难题。
技术实现要素:
本发明首先提供一种药物中间原料,有效提高了以当归和川芎为原料制备的挥发油的稳定性。
一种药物中间原料,包括挥发油和环糊精,其中,所述环糊精将所述挥发油完全包合或基本上完全包合;所述挥发油是以当归和川芎为原料制备得到的。
根据本发明实施例,所述环糊精为β-环糊精。
根据本发明实施例,所述挥发油和环糊精的重量比为1:5-8。
实验证明,选用该比例的环糊精可将所述挥发油完全包合或基本上完全包合。经包合后的挥发油显著地提高了稳定性,所述药物中间原料中挥发油的含量及组成未发生显著变化。
本发明中,所述完全包合的含义是指被包合的挥发油各组分全部进入到环糊精的空穴中,理论包封率为100%。
本发明中,所述基本上完全包合的含义是指挥发油经环糊精包合后通过感官识别不到挥发油颜色及气味,且测定包封率不低于90%。
本发明中,所述包封率是指被包封的挥发油占总挥发油的质量百分比;一般以包合物中挥发油的量与挥发油的加入量及挥发油含量测定的空白回收率进行计算表征。
根据本发明实施例,所述药物中间原料的包封率≥90。
根据本发明实施例,制备所述挥发油的原料中,当归和川芎的重量比为1-5:1-5,优选为1-3:1-3,更优选为3:2。
根据本发明实施例,所述挥发油是以当归和川芎为原料采用水蒸气蒸馏法制备得到的。一般地,加水量为当归和川芎重量的4-8倍;通常可浸泡6-24h再进行蒸馏。蒸馏时间通常为2-6h。
本发明还提供上述药物中间原料的制备方法,包括:
提供所述挥发油;
提供所述环糊精;
将所述环糊精和所述挥发油进行包合。
根据本发明实施例,是采用研磨法进行包合的,即将所述环糊精和所述挥发油按照优化的比例,通过手动研磨或球磨机进行研磨包合至所述挥发油的颜色及气味被完全或基本上完全被包合为止。
根据本发明实施例,在包合前可先将所述环糊精用水混合分散均匀。通常,所述环糊精与水的重量比为1:1-5,例如1:1.5-2。
根据本发明实施例,在包合前可先将所述挥发油用无水乙醇稀释。通常,所述挥发油与无水乙醇的重量比为1:1-2。通过适量无水乙醇稀释可以增加挥发油与环糊精的有效接触面积,利于挥发油分子进入到环糊精空穴,提高包合效率。
根据本发明实施例,可先将当归和川芎药材或饮片破碎后,再提取挥发油。例如通常可破碎至5-15mm。
在本发明一些具体实施例中,所述挥发油的制备方法包括:取当归和川芎药材或饮片,破碎,用4-8倍量水浸泡6-24h,采用水蒸气蒸馏法提取挥发油,提取时间为2-6h,收集挥发油。在一些实施例中,还可进一步收集蒸馏液,其用途可作为制备中药组合物,具体可参见下文。
在本发明一些具体实施例中,所述药物中间原料的制备方法包括:
取所述挥发油,用1-2倍重量的无水乙醇溶解稀释;
取所述环糊精,用1-5倍重量的水混合分散均匀;
将以上稀释后的挥发油和环糊精混合,优选控制所述挥发油和环糊精的重量比为1:5-8,进行研磨,研磨至所述挥发油的颜色及气味被完全或基本上完全被包合为止。
在本发明一些具体实施例中,还包括将包合后包合物进行冷藏处理的步骤;所述冷藏处理的温度优选为0-5℃,冷藏时间优选为12-24h。
研究过程中发现,包合物形成后至干燥前,仍处于动态的不稳定状态。例如,采用抽滤或离心的方式除水过程中,部分挥发油分子因受外力会从环糊精空穴中迁移出,而在接下来的加热干燥过程进一步迁移除。发明人经过研究发现,通过冷藏,例如具体可在0-5℃冷藏12-24h,可以提高包合率。分析原因可能为,冷藏可增强挥发油分子与β-环糊精的络合能力,提高挥发油分子抵抗抽滤或离心产生的抽离力;同时,通过冷藏还可以降低环糊精在水中的溶解,减少一部分包合物因溶解在水中而造成的损失,提高包合物收率。
在本发明一些具体实施例中,还包括在上述冷藏处理后进行除水处理的步骤,即除去包合物中的水分或部分水分。可采用本领域常规除水方法,例如离心除水。
在本发明一些具体实施例中,还包括在上述除水处理后进行干燥的步骤。可采用本领域常规干燥方法,例如45℃条件下减压干燥。
环糊精是一种环状低聚多糖,无毒、可食用,具有外缘亲水而内腔疏水略呈锥形的中空环状结构,其环内空间正好可容纳挥发油分子,在一定条件下挥发油分子进入环糊精空腔形成包合物,受环糊精空腔的保护,挥发油类成分能有效避免空气中氧气的氧化、遇光后的分解和挥发。本发明挥发油经β-环糊精包合后,稳定性较挥发油与环糊精的混合物相比显著提高。
本发明还包括上述方法制备的药物中间原料。
本文中,所述药物中间原料有时也称为挥发油包合物。
本发明上述药物中间原料可作为制备中药组合物或药物制剂的原料。
本发明还提供一种中药组合物,其主要成分包括上述药物中间原料,还包括提取物;其中,所述提取物是以当归和川芎为原料制备得到的。
根据本发明实施例,所述提取物的含义也包括液态的提取物,例如提取液。
根据本发明实施例,所述提取物的含义不包括上述挥发油。
根据本发明实施例,制备所述提取物的原料中,当归和川芎的重量比为1-5:1-5,优选为1-3:1-3,更优选为3:2。
根据本发明实施例,所述提取物的制备方法包括:
1)取当归和川芎药材或饮片,破碎,采用水蒸气蒸馏法提取挥发油,收集挥发油和蒸馏液;
2)向步骤1)提取挥发油后的物料加入乙醇继续提取,得醇提取液;
3)将步骤2)所得醇提取液低温静置,离心,将所得上清液采用减压方式进行浓缩,得浓缩液;
4)将步骤1)所得蒸馏液和步骤3)所得浓缩液合并,澄清,离心,将所得上清液采取减压方式除去溶剂,即得所述提取物。
进一步地,步骤1)具体方法可参见上文所述挥发油的制备方法。
进一步地,步骤2)所用乙醇的浓度为50-100%,可选为85-95%,例如85%或90%。通常可用乙醇提取1-3次,例如1次、2次或3次。每次所用乙醇的量可为当归和川芎药材重量的4-10倍。每次提取以药液沸腾一次后静置6-12h为宜。也就是说,在每次提取将药液沸腾后静置6-12h,再进行提取。研究发现,采用这种煮沸一次即停止加热,静置至室温放出药液的提取方式,可以确保有效成分阿魏酸的稳定性。步骤2)中,在乙醇加入的瞬间,水提取液中产生大量白色絮状漂浮物(为杂质),此漂浮物可经药渣截留,提取过程伴随除杂。
进一步地,步骤3)中将步骤2)所得醇提取液可先低温静置一段时间,例如静置8-24小时,再离心,所述低温一般是指0-10℃。通过静置可以析出白色絮状杂质,达到除杂的目的。
进一步地,步骤3)中,可采用减压方式进行浓缩,例如通常可在40-60℃减压回收溶剂,浓缩至每1ml药液含0.2-1.0g生药的浓缩液。
进一步地,步骤4)中,向合并液中加入壳聚糖溶液进行澄清,所述壳聚糖加入量为合并液总质量的8-15%,例如10%。所述壳聚糖溶液的浓度为0.2-2%,例如1%。加入壳聚糖溶液后,搅拌均匀,静置,离心,以进一步除去絮状杂质。
进一步地,步骤4)中,上清液采取减压方式除去溶剂,例如可在40-55℃减压回收溶剂,浓缩至每1ml药液含3.5-5.5g生药的浓缩液。
在本发明一些具体实施例中,所述提取物的制备方法包括:
1)取当归和川芎药材或饮片,破碎,用4-8倍量水浸泡6-24h,采用水蒸气蒸馏法提取挥发油,提取时间为2-6h,收集挥发油和蒸馏液;
2)向步骤1)提取挥发油后的物料(药渣药液)中加入4-8倍重量浓度为50-100%乙醇,加热至煮沸,停止加热,静置至室温,分离出提取液;再次加入6-10倍重量50-100%乙醇,加热至药液沸腾,停止加热,药液温度降室温,收集提取液,合并两次药液,得醇提取液;
3)将步骤2)所制得醇提取液,低温(0-10℃)静置8-24h,离心,除去析出物,上清液40-60℃减压浓缩至每1ml药液含0.2-1.0g的生药,得醇提物浓缩液;
4)合并步骤1)所得蒸馏液与步骤3)所得醇提物浓缩液,加入总质量8%-15%的浓度为0.2-2%的壳聚糖溶液,搅拌均匀,室温静置8-24h,离心,40-55℃减压回收溶剂,浓缩至每1ml药液含3.5-5.5g生药的浓缩液,即为提取物。
在本发明一些更具体实施例中,所述提取物的制备方法包括:
1)取当归和川芎药材或饮片,破碎,用6倍量水浸泡12h,采用水蒸气蒸馏法提取挥发油,提取时间为5h,收集挥发油和蒸馏液;
2)向步骤1)提取挥发油后的物料中加入8倍重量浓度为90%乙醇,加热至煮沸,停止加热,静置至室温,分离出提取液;再次加入8倍重量90%乙醇,加热至药液沸腾,停止加热,药液温度降室温,收集提取液,合并两次药液,得醇提取液;
3)步骤2)所制得醇提取液,低温(0-5℃)静置24h,离心,除去析出物,上清液45-55℃减压浓缩至每1ml药液含0.2-1.0g的生药,得醇提物浓缩液;
4)合并步骤1)所得蒸馏液与步骤3)所得醇提物浓缩液,加入总质量10%的浓度为1%的壳聚糖溶液,搅拌均匀,室温静置24h,离心,45-50℃减压回收。
本发明所述方法制得的提取物纯度高,杂质含量低,膏体不粘稠、利于干燥及后续制剂的生产。本发明依据“佛手散”原方制法,采取水提加醇提的方式制备总提取物,在水蒸气蒸馏法提取挥发油的基础上,向药渣液中加入乙醇继续提取,意外的发现,乙醇加入后产生大量白色的絮状漂浮物,此絮状物可通过药渣有效的截留,即在提取的过程中伴随除杂,收集到的提取液杂质含量大幅降低。
本发明还提供一种中药制剂,其含有上述中药组合物,或者还进一步含有药学可接受的辅料。
根据本发明实施例,所述中药制剂可由上述中药组合物单独制成;或者进一步与药学可接受的辅料一起制成。
根据本发明实施例,所述辅料可选自淀粉、糊精、糖粉、微晶纤维素、乳糖、甘露醇、薄荷脑、维生素c、柠檬酸、二氧化硅、生育酚、麦芽糖醇、蔗糖脂肪酸酯、阿巴斯甜、硬脂酸镁、交联羧甲基纤维素钠、羟甲基纤维素、羟丙纤维素、聚维酮、聚丙烯酸树脂ⅳ,聚乙二醇中的一种或几种。
根据本发明实施例,所述中药制剂由上述药物中间原料(挥发油包合物)及上述提取物的干膏粉制成。
根据本发明实施例,所述中药制剂由上述药物中间原料(挥发油包合物)及所述提取物或所述提取物的浓缩液制成。具体例如,将所述药物中间原料(挥发油包合物)加入到所述提取物的浓缩液中,或将所述药物中间原料(挥发油包合物)连同辅料一起与所述提取物浓缩液进行混合或制粒。其优势在于,既实现了全组分的均匀混合,又省去提取物的干燥步骤。
根据本发明实施例,所述中药制剂包括胶囊剂、颗粒剂、胶囊剂、片剂。
本发明药物中间原料的制备采用研磨法包合,为物理捏合,包合率高,包合前后挥发油成分组成未发生变化,操作简便、无环境污染,易于工业化生产,易于推广应用。本发明提供的提取物采用的物理方法对水溶性浸膏进行除杂处理,解决了存在于中药水溶性浸膏杂质含量高、膏体粘稠、不利于干燥及后续制剂的生产的问题。本发明提供的中药制剂保持了佛手散原方物质基础,提供了一种供口服的固体制剂,能够更好地发挥原方养血活血、去瘀生新的功能,具有较好的临床应用价值和市场推广价值。
附图说明
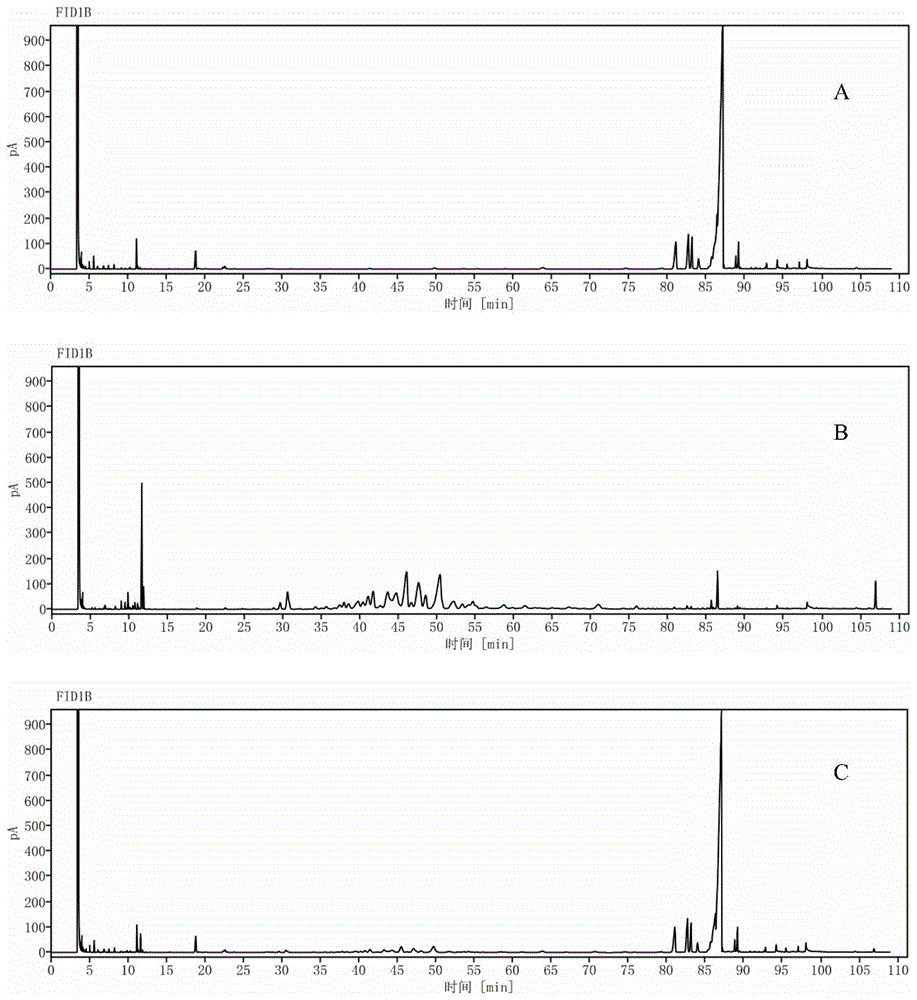
图1为实验例1样品在高温60℃条件下放置10天的气相图谱。
图2为实验例1样品在光照4500lx±500lx条件下放置10天的气相图谱。
图3为实验例2样品的气相图谱。
图1-3中,纵坐标中pa表示响应值,fid1b表示氢火焰离子检测器。
图4为实验例3样品以正己烷-乙酸乙酯(9:1)为展开系统的tlc图谱。
图5为实验例3样品以环己烷-二氯甲烷-乙酸乙酯-甲酸(4:1:1:0.1)为展开系统的tlc图谱。
图6为实验例3样品以正丁醇-冰醋酸-水(4:1:1)为展开系统的tlc图谱。
具体实施方式
以下通过实施例详细说明本发明技术方案,并不以此限定本发明的实施范围。
实施例1挥发油的制备
将当归、川芎破碎为5-15mm的粗颗粒,当归与川芎按重量比3:2投料,加入6倍量水浸泡24h、水蒸气蒸馏提取挥发油5h,分别收集挥发油与蒸馏液。挥发油收率为0.52%(ml/g)。
实施例2提取物的制备
向实施例1提取挥发油后的物料(药渣药液)中,加入8倍重量90%乙醇,加热至药液沸腾,停止加热,温度降至室温,自提取罐底部缓慢收取提取液。药渣中再次加入8倍重量90%乙醇,加热至药液沸腾,停止加热,温度降至室温,自提取罐底部缓慢收取提取液,合并两次提取液,得醇提取液;0-5℃条件下静置24h,离心,得上清液;45-50℃减压回收溶剂,浓缩至0.4g生药/ml药液,得醇提物浓缩液;将醇提取物浓缩液与实施例1所制得的蒸馏液合并,加入总质量10%浓度为1%的壳聚糖溶液,搅拌均匀,室温静置24h,离心,45-50℃减压回收溶剂,浓缩至每1ml药液含2.5g生药的浓缩液,即为提取物。
实施例3挥发油包合物的制备
取实施例1所制挥发油,加入2倍重量无水乙醇溶解稀释,得挥发油乙醇溶液,备用。
称取实施例1所制挥发油8倍重量β-环糊精,将β-环糊精与水按1:2重量比例投入球磨机中研磨至均质;向球磨机中加入上述挥发油乙醇溶液,启动球磨机进行包合,至无挥发油颜色及气味,物料呈粘稠状糊状物;出料,0-5℃冷藏24h;低速离心除水;包合物于45℃条件下减压干燥,得挥发油包合物。
本实施例制备的挥发油包合物可作为药物中间原料,进一步用于制备药物制剂。
取本实施例制备的干燥后的挥发油包合物,经无水乙醇抽滤淋洗3次,45℃减压干燥,得洗涤后包合物,按2020版《中国药典》四部<挥发油含量测定法-乙法>测定洗涤后包合物挥发油含量及空白回收率,计算包封率为95.7%。
实施例4胶囊剂制备
取实施例3制备的挥发油包合物和实施例2制备的的提取物,备用;
将该提取物经45-50℃减压浓缩至3.5g生药/ml药液,进一步于50-55℃减压干燥,得提取物干膏粉。
取该提取物干膏粉,粉碎、过筛,与实施例3制备的挥发油包合物及适量二氧化硅混合均匀,灌装胶囊,即得胶囊剂。
实施例5颗粒剂制备
按实施例1-3相同的方法制备挥发油、提取物和挥发油包合物。
将该提取物经45-50℃减压浓缩至3.5g生药/ml药液,得提取液浓缩液;
将该挥发油包合物与适量柠檬酸、麦芽糖醇、糖粉和糊精,混合均匀,加入至所述提取液浓缩液中,混匀,50℃减压干燥,得中间体颗粒;
将该中间体颗粒,过筛整粒,加入适量二氧化硅,混合均匀,即得颗粒剂。
实施例6片剂制备
按实施例1-3相同的方法制备挥发油、提取物和挥发油包合物。
将该提取物经45-50℃减压浓缩,至4.5-5.0g生药/ml药液,得浓缩后的提取物;
将微晶纤维素、预胶化淀粉、所述挥发油包合物、维生素c、蔗糖脂肪酸酯及交联羧甲基纤维素钠依次加入至湿法混合制粒机中,混合;加入进一步所述浓缩后的提取物,低速制粒,湿颗粒于50-60℃减压干燥,得中间体颗粒;将该中间体颗粒,16目圆形筛整粒,加入适量硬脂酸镁,混合均匀,压片,包薄膜衣,即得。
实验例1挥发油经环糊精包合后稳定性考察
试验样品:实施例3制备的挥发油包合物。
对照样品:按实施例1相同的方法制备挥发油;将该挥发油用2倍重量的无水乙醇溶解稀释,得挥发油乙醇溶液;加入挥发油8倍重量的β-环糊精,混匀,40℃减压干燥,即得。
试验方法:分别将以上样品置于高温60℃、光照4500lx±500条件下放置10天,考察挥发油含量变化。
检测方法:挥发油含量测定-参照2020年版《中国药典》四部<挥发油测定法-乙法>挥发油组成检测-气相色谱法,(5%-苯基)-甲基聚硅氧烷毛细管柱,fid检测器,载气:氮气,载气流量:1.5ml/min。程序升温:初始温度90℃,以每分钟10℃的速率升温至120℃,保持70分钟,以每分钟5℃的速率升温至250℃,保持10分钟。进样口温度为250℃,检测器温度为260℃,分流比为10:1。
样品处理:试验样品与对照样品均经无水乙醇超声提取,过滤,测定气相图谱。
试验结果:
(1)高温60℃条件下放置10天
结果见下表1和图1。图1是挥发油(样品a,见实施例1)、对照样品(60℃,放置10天,样品b)、试验样品(60℃,放置10天,样品c)的气相图谱(gc图谱)。
表1高温条件挥发油含量测定结果
由表1可知,高温60℃条件下,对照样品中挥发油损失严重,含量下降约85%,由gc图谱可知,对照样品主峰相对含量显著降低,且新增大量杂峰,表明挥发油组分在持续高温下降解、挥发;而形成包合物后挥发油含量下降15%,gc图谱显示略有降解,但整体组成未发生显著变化,与对照样品相比,稳定性显著改善。
(2)光照4500lx±500lx条件下放置10天
结果见下表2和图2。图2是挥发油(样品a,见实施例1)、对照样品(4500lx±500lx,放置10天,样品b)、试验样品(4500lx±500lx,放置10天,样品c)的气相图谱。
表2光照条件挥发油含量测定结果
表2显示,4500lx条件下放置10天,对照样品中挥发油含量发现了显著变化,含量下降26%,包合物中挥发油含量几乎未发生改变;同时,由气相图谱(gc图谱)可知,对照样品挥发油组成亦发生显著变化,包合物中挥发油组成未发生显著变化,与未包合的原油基本一致。说明,挥发油经环糊精包合后,光照稳定性显著提高。
以上结果表明,本发明成功对挥发油进行包合,且包合物耐受光照、耐受高温的能力显著提升,有效解决了挥发油类有效成分稳定性差的问题。
实验例2组合物中挥发油测定
采用气相色谱法对本发明提供的制剂进行挥发油测定。
试验样品:实施例4制备所得胶囊剂(样品a);
实施例5制备所得颗粒剂(样品b);
实施例6制备所得片剂(样品c)。
对照样品:实施例1制备所得挥发油(样品d)。
实验方法:气相色谱法,参数同实验例1。
样品处理:对照样品-挥发油经无水乙醇溶解稀释,即得。
胶囊剂取内容物,经无水乙醇超声提取,过滤,即得。
颗粒剂经无水乙醇超声提取,过滤,即得。
片剂研成细粉后,经无水乙醇超声提取,过滤,即得。
试验结果见图3。图3为以上样品的气相图谱(gc图谱)。
由图3可知,本发明提供的胶囊剂、颗粒剂及片剂中挥发油成分组成与未经包合的挥发油基本一致。说明本发明在对挥发油进行包合后,并经进一步制成制剂的过程中,挥发油组成未发生变化,为挥发油类活性成分的临床应用提供了有效解决方案。
实验例3本发明提取物与“佛手散”煎液化学成分对比分析
对照样品:按“佛手散”原方制法制备煎液,具体为当归、川芎粉碎成过20目筛的粗粉,随即按3:2比例混合,加5倍量水于电磁炉煎煮,至药液液面下降至露出药渣,加入8倍量90%乙醇,至药液再次沸腾,停止加热,药液降至室温时,滤除药渣,得提取液,即得佛手散煎液。
成分对照:阿魏酸(阿魏酸为当归、川芎两味药材发挥药效活性的标志性成分,故以阿魏酸作为对照分析煎液和提取物。)
试验样品:实施例1所制挥发油;实施例2所制提取物。
试验方法:薄层色谱法,采用不同展开系统对样品各组分进行层析考察
试验结果:
(1)以正己烷-乙酸乙酯(体积比9:1)为展开系统,进行小极性成分分析,结果见图4。图4中,样1为对照样品(佛手散煎液)、样2为实施例1所制挥发油、样3为实施例2所制提取物。
图4为鉴别挥发油类小极性成分的展开系统下所得到的tlc图谱。由图4可见本发明所制挥发油与提取物因分段提取斑点组成有所不同,但整体对比,二者与佛手散煎液主斑点组成基本一致。图4中挥发油显示的斑点信息较多,因其浓度与纯度较高点样量较大所致。
(2)以环己烷-二氯甲烷-乙酸乙酯-甲酸(体积比4:1:1:0.1)为展开系统,进行中、高极性成分分析,结果见图5。
图5中,样1为对照样品(佛手散煎液)、样2为阿魏酸、样3为实施例2所制提取物。
图5为鉴别特征成分阿魏酸的展开剂所层析得到的tlc图谱,由图5可知,对照样品佛手散煎液与本发明提取物中均含有阿魏酸,且二者tlc斑点组成未见差异。
(3)以正丁醇-冰醋酸-水(体积比4:1:1)为展开系统,对高极性成分进行分析,结果见图6。图6中,样1为对照样品(佛手散煎液)、样2为阿魏酸、样3为实施例2所制提取物。由图6可知,对照样品佛手散煎液与本发明提取物高极性段tlc斑点组成一致。
图4、图5、图6表明本发明所制组合物成分组成与佛手散煎液基本一致。
虽然,上文中已经用一般性说明、具体实施方式,对本发明作了详尽的描述,但在本发明基础上,可以对之作一些修改或改进,这对本领域技术人员而言是显而易见的。因此,在不偏离本发明精神的基础上所做的这些修改或改进,均属于本发明要求保护的范围。
- 还没有人留言评论。精彩留言会获得点赞!