用于有不良渗透性的蛋白质、肽和小分子的口服递送的制剂的制作方法
用于有不良渗透性的蛋白质、肽和小分子的口服递送的制剂
1.相关申请的交叉引用本技术要求于2019年4月11日提交的美国临时申请号62/832,508的优先权,其全部内容通过引用并入本文。
2.公开领域本公开涉及用于有不良渗透性的蛋白质、肽和小分子的口服递送的制剂。更具体地,本公开涉及意图用于任意有治疗活性的有不良渗透性的合成或天然分子或其盐或溶剂化物的口服递送的药物制剂。
3.发明背景不良渗透性分子为通过肠膜的吸收不良的化合物。因此,它们被静脉内或皮下施用。由于它们通过肠膜的吸收不良,鉴于静脉内施用和每天多次给药的需要(例如,胰岛素对于糖尿病患者),它们的临床应用显著受限。这些不良渗透性化合物在由amidongl等人于atheoreticalbasisforabiopharmaceuticdrugclassification:thecorrelationofinvitrodrugproductdissolutionandinvivobioavailability(pharmres.1995mar;12(3):413-20.)中提出的分类中被确定为bcsiii类和iv类化合物,所述文件以其全文通过引用并入本文。
4.发明概述申请人已经开发了用于有不良渗透性的口服施用分子的制剂。所述分子可为cgrp抑制剂。这些制剂非常有益于需要每天多次给药的患者。为了制备用于不良渗透性cgrp抑制剂的口服递送的此类制剂,申请人必须至少克服:相对于肠膜的此不良渗透性;及对于那些抑制剂中的一些,特别是肽和蛋白质,在胃肠道中的化学和物理不稳定性,且特别是由于在胃中的酸性环境而失去活性;以及整个肠中的酶促降解。相应地,申请人开发了延迟释放包衣剂型,其能在肠中递送不良渗透性cgrp抑制剂,并且原位生产渗透促进剂以提高其生物利用度。
5.在美国专利号9,259,389中,发明人发现可消化反相乳液能提高低聚糖的生物利用度。意外的是,申请人发现有不良渗透性分子作为粉末分散在制剂中的脂质赋形剂溶液对于此特定类分子(即在由amidongl等人(pharmres.1995mar;12(3):413-20.)提出的分类中的bcsiii类和iv类化合物)可容许更好的生物利用度结果。特别地,申请人发现对于不良渗透性分子,特别是bcsiii类蛋白质和肽化合物,不加入水的制剂可为有益的。不受任何理论限制,相信水趋向于导致此类不良渗透性分子聚集在一起。更特别的是,申请人发现,当他们在有不良渗透性bcsiii类蛋白质或肽分子或盐作为粉末分散在制剂中的包含脂质基赋形剂溶液的制剂中不包括水时,实现了对于此特定类分子的更高生物利用度的结果。相反,对于美国专利号9,259,389中的糖类,水的去除是有害的。
6.另外,当活性药物成分(“api”)可被作为粉末分散而不需要在水中溶解api时,鉴于不需要溶解api,申请人可增加药物载量。此外,该制剂本质上更物理稳定,因为脂质赋形剂可在溶液中作为单相。因此可不需要添加例如二氧化硅的稳定剂来稳定各相。在一些实施方案中,出于制造目的,可加入增稠剂以维持在过程中api粉末在悬浮液中的均匀性。在
一些实施方案中,增稠剂可为二氧化硅。最后,相比于文献中发现的使用比如渗透促进剂的赋形剂的其他制剂,本文公开的制剂可使用仅普遍公认为安全的赋形剂或已投入市场的成分。
7.在一些实施方案中,药物制剂包含:制剂总重量的0.01-20重量%的量的合成或天然的不良渗透性cgrp抑制剂或其盐或溶剂化物;制剂总重量的50-80重量%的量的包含脂肪酸的甘油三酯的亲油相;以及制剂总重量的10-50重量%的量的至少一种包含多元醇与脂肪酸的偏酯的亲油表面活性剂。在一些实施方案中,合成或天然的不良渗透性cgrp抑制剂或其盐或溶剂化物为cgrp抗体、cgrp受体抗体、来自cgrp抗体或cgrp受体抗体的抗原结合片段、cgrp输注抑制蛋白、cgrp生物中和剂、小分子cgrp受体拮抗剂、小分子cgrp抑制剂或多肽cgrp抑制剂。在一些实施方案中,小分子cgrp受体拮抗剂为(r)-n-(3-(7-甲基-1h-吲唑-5-基)-1-(4-(1-甲基哌啶-4-基)哌嗪-1-基)-1-氧代丙-2-基)-4-(2-氧代-1,2-二氢喹啉-3-基)哌啶-1-甲酰胺 (bhv-3500)。在一些实施方案中,制剂包含至少一种亲水亲油平衡(“hlb”)高于10的亲水表面活性剂,其占制剂总重量的1-30重量%的量。在一些实施方案中,所述至少一种亲水表面活性剂选自:聚氧乙烯(20)单油酸酯、peg 8 辛酸/癸酸甘油酯、peg 6 辛酸/癸酸甘油酯、聚(氧乙烯)(4)月桂醚和它们的混合物。在一些实施方案中,脂肪酸的甘油三酯为中链脂肪酸。在一些实施方案中,亲油表面活性剂包含中链脂肪酸的甘油单酯和甘油二酯的混合物。在一些实施方案中,制剂不包括水。在一些实施方案中,延迟释放药物剂型包含以上描述的任意制剂,其中延迟释放剂型为包衣剂型,其释放由ph决定。在一些实施方案中,用于治疗患者的方法包含将有效量的任意上述制剂施用给需要其的人。
8.从以下详细描述,对于本领域技术人员而言另外的优点将容易显而易见。本文的例子和描述应视为本质上说明性的而非限制性的。
9.所有本技术提及的出版物,包括专利文件、科学文章和数据库,都出于所有目的通过引用以其全文并入,达到如同每个单独出版物通过引用单独并入的相同程度。如果本文提出的定义与通过引用并入本文的专利、申请、公开申请及其他出版物所提出的定义相反或以其他方式不一致,则本文提出的定义优先于通过引用并入本文的定义。
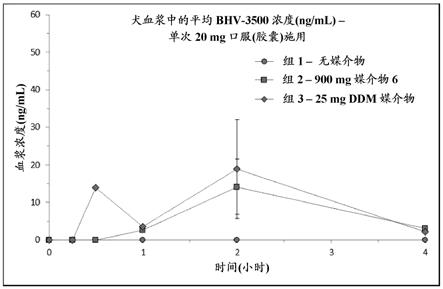
10.附图简要说明通过以下结合附图对实施方案的描述,这些和/或其他方面将变得显而易见并更容易理解,在附图中:图1为曲线图,针对组1至3 (胶囊;20mg)示出了犬血浆中的平均bhv-3500浓度分布曲线;和图2为曲线图,针对组4至6 (胶囊;50mg)示出了犬血浆中的平均bhv-3500浓度分布曲线。
11.发明详细描述本公开涉及意图用于口服施用的药物制剂,其包含合成或天然的不良渗透性分子且有治疗活性或其药学上可接受的加成盐或溶剂化物。这些制剂可为脂质基制剂。另外,这些制剂可为延迟释放剂型。在一些实施方案中,所述剂型可为延迟释放软凝胶胶囊、硬壳胶囊或其组合。在一些实施方案中,此延迟释放剂型可为肠溶释放剂型。
12.制剂可包括:(a)合成或天然的不良渗透性分子;(b)亲油相;(c)至少一种亲油表面活性剂;和/或(d)至少一种亲水表面活性剂。在一些实施方案中,制剂可包括化学和/或
物理稳定剂。
13.合成或天然的不良渗透性分子 在一些实施方案中,制剂可包括合成或天然的不良渗透性分子或这些不良渗透性分子的任意药学上可接受的盐,其占制剂总重量的至多约1重量%、约2重量%、约5重量%、约10重量%、约15重量%或约20重量%的量。在一些实施方案中,制剂可包括合成或天然的不良渗透性分子或这些不良渗透性分子的任意药学上可接受的盐,其占制剂总重量的约0.01-30重量%、约0.1-30重量%、约0.01-20重量%、约0.1-20重量%、约0.1-15重量%、约0.1-10重量%、约0.1-5重量%、约0.1-2重量%、约0.1-1重量%、约0.1-0.5重量%或约0.5-1.5重量%的量。
14.合成或天然的不良渗透性分子或其药学上可接受的盐可包括:意图用于口服递送的有不良渗透性的任意蛋白质、多肽、肽或小分子,其中根据本发明的活性组分可为但不限于胰岛素、人生长激素、降钙素(例如鲑鱼降钙素)、干扰素比如α-、β-或γ-干扰素、胰高血糖素、促性腺激素释放激素、脑啡肽、疫苗、酶、激素类似物、酶抑制剂、抗体和抗体模拟物。合成或天然的不良渗透性分子或其药学上可接受的盐为由amidongl等人于atheoreticalbasisforabiopharmaceuticdrugclassification:thecorrelationofinvitrodrugproductdissolutionandinvivobioavailability(pharmres.1995mar;12(3):413-20.)中提出的分类中被确定为bcsiii类和iv类的那些。
15.cgrp抑制剂合成或天然的不良渗透性分子可为降钙素基因相关肽(cgrp)抑制剂。如本文所用,术语“cgrp抑制剂”是指可为cgrp配体或cgrp受体的抑制剂的化学实体。因此,术语“cgrp抑制剂”涵盖cgrp受体抑制剂。cgrp抑制剂可为cgrp抑制剂或cgrp受体抑制剂。cgrp(降钙素基因相关肽)为一种37个氨基酸的神经肽,其属于包括降钙素、肾上腺髓质素和胰淀素在内的肽家族。已收集到大量证据表明cgrp与偏头痛的病理生理学有关。进行了临床试验以证明cgrp抑制剂对治疗偏头痛有效。
16.cgrp抑制剂可为cgrp抗体、cgrp受体抗体、来自cgrp抗体或cgrp受体抗体的抗原结合片段、cgrp输注抑制蛋白、cgrp生物中和剂、小分子cgrp受体拮抗剂、小分子cgrp抑制剂或多肽cgrp抑制剂。在一个实施方案中,cgrp抑制剂可为小分子cgrp受体拮抗剂。小分子cgrp受体拮抗剂可为(r)-n-(3-(7-甲基-1h-吲唑-5-基)-1-(4-(1-甲基哌啶-4-基)哌嗪-1-基)-1-氧代丙-2-基)-4-(2-氧代-1,2-二氢喹啉-3-基)哌啶-1-甲酰胺(bhv-3500)。
17.cgrp抑制剂可以每天约1-1000mg的剂量施用。在另一个方面,cgrp抑制剂以每天约1、5、10、15、20、25、30、40、50、60、70、80、90、100、200、250、300、400、500、750或1000mg的剂量施用。cgrp抑制剂的日剂量可在任意上述值之间的范围内。
18.亲油相在一些实施方案中,制剂可包括亲油相,其占制剂总重量的至多约50重量%、约55重量%、约60重量%、约65重量%、约70重量%或约80重量%的量。在一些实施方案中,制剂可包括亲油相,其占制剂总重量的约50-80重量%、约55-75重量%、约60-70重量%、约62-68重量%、约64-66重量%或约65重量%的量。
19.在一些实施方案中,亲油相可为脂肪酸的甘油三酯。脂肪酸的甘油三酯可指任意药学上和口服可接受的饱和或不饱和脂肪酸的甘油三酯。在一些实施方案中,脂肪酸的甘
油三酯可有下式:其中r1、r2和r3彼此独立地代表母体脂肪酸的烷基或烯基。
20.脂肪酸可为饱和的或不饱和的。特别地,脂肪酸可为饱和的,因为不饱和脂肪酸可导致更慢的消化动力学和更低的消化百分比。一些常见的饱和脂肪酸在以下表1中指出。
21.表1r1、r2和r3可代表直链或支链。在一些实施方案中,r1、r2和r3可为c3-c23烷基或烯基、c5-c13烷基或烯基或者c7-c9烷基或烯基。在一些实施方案中,脂肪酸为饱和脂肪酸且为中链脂肪酸。因此,亲油相可为长(比如豆油和鱼油)、中或短(比如三乙酸甘油酯)链脂肪酸的甘油三酯。在一些实施方案中,甘油三酯可为辛酸、癸酸或其混合物的甘油三酯(比如商业产品miglyol 812
®
、captex 355
®
、estasan
®
、neobee m5
®
、labrafac cc
®
和captex 1000
®
)。在一些实施方案中,甘油三酯可为c6-c12脂肪酸或c8-c10脂肪酸的甘油三酯。
22.亲油表面活性剂在一些实施方案中,制剂可包括至少一种亲油表面活性剂,其占制剂总重量的至多约1重量%、约5重量%、约10重量%、约15重量%、约20重量%、约25重量%、约30重量%、约35重量%、约40重量%、约45重量%或约50重量%的量。在一些实施方案中,制剂可包括至少一种亲油表面活性剂,其占制剂总重量的约10-50重量%、约15-35重量%、约20-30重量%、约22-28重量%、约24-26重量%或约25重量%的量。如果制剂包括少于约10重量%的至少一种亲油表面活性剂,动力消化可能未被优化。如果制剂包括超过50重量%的至少一种亲油表面活性剂,可
用于释放癸酸钠的亲油相的量可能不为最优。
23.在一些实施方案中,所述至少一种亲油表面活性剂可为多元醇与脂肪酸的偏酯。多元醇与脂肪酸的偏酯可指药学上和口服可接受的由多元醇和饱和或不饱和脂肪酸的酯化作用得到的任意偏酯。常见的饱和脂肪酸在以上提到的表1中指出。脂肪酸可为中链脂肪酸,比如c6-c12 脂肪酸,特别是辛酸和/或癸酸。多元醇可为诸如丙二醇和甘油。比如,多元醇与脂肪酸的偏酯可为丙二醇脂肪酸单酯和/或二酯(比如以商品名lauroglycol
®
出售的丙二醇单月桂酸酯、以商品名mirpyl
®
出售的丙二醇单肉豆蔻酸酯或以商品名captex 200
®
、miglyol 840
®
或neobee m-20
®
出售的丙二醇二辛酸酯/二癸酸酯)和/或脂肪酸的聚甘油酯(比如以商品名plurol oleique
®ꢀ
或 drewpol 10.10.10
®
出售的聚甘油油酸酯或以商品名caprol et
®
出售的聚甘油混合脂肪酸)。
24.至少一种亲油表面活性剂可为丙二醇和脂肪酸的偏酯(比如商业产品capryol pgmc
®ꢀ
和capmul pg-8
®
)。在一些实施方案中,至少一种亲油表面活性剂可为脂肪酸甘油单酯和甘油二酯的混合物、中链脂肪酸甘油单酯和甘油二酯的混合物、辛酸和/或癸酸甘油单酯和甘油二酯的混合物(比如商业产品capmul mcm 和capmul mcm c8
®
、imwitor 988
®
、imwitor 742
®
)或癸酸甘油单酯和甘油二酯的混合物(比如商业产品capmul mcm c100或imwitor 308
®
)。
25.在一些实施方案中,至少一种亲油表面活性剂可为卵磷脂,例如大豆卵磷脂,如但不限于大豆卵磷脂。
26.亲水表面活性剂在一些实施方案中,制剂可包括至少一种亲水表面活性剂,其占制剂总重量的至多约2重量%、约5重量%、约8重量%、约10重量%、约15重量%、约20重量%、约25重量%或约30重量%的量。在一些实施方案中,制剂可包括至少一种亲水表面活性剂,其占制剂总重量的约0-30重量%、约0-15重量%、约0-10重量%、约1-30重量%、约5-15重量%、约8-12重量%、约9-11重量%或约10重量%的量。如果至少一种亲水表面活性剂的量大于制剂的约30重量%,可用于释放癸酸钠的亲油相的量可被折衷。
27.在一些实施方案中,至少一种亲水表面活性剂可为药学上和口服可接受的亲水亲油平衡(“hlb”)值高于10的任意亲水表面活性剂。hlb值为本领域技术人员常用以表征非离子表面活性剂的相对亲水性和疏水性的经验参数。
28.在一些实施方案中,至少一种亲水表面活性剂可为:磷脂;聚氧乙烯脱水山梨糖醇脂肪酸衍生物,比如聚氧乙烯(20)单月桂酸酯(以商品名吐温 20
®
出售)、聚氧乙烯(20)单油酸酯(以商品名吐温 80
®ꢀ
和/或crillet 4
®
出售)或聚氧乙烯(20)单棕榈酸酯(以商品名montanox 40
®
出售);hlb值高于10的蓖麻油或氢化蓖麻油乙氧基化物,比如聚氧乙烯(35)蓖麻油(以商品名cremophor el
®
出售),聚氧乙烯(40)氢化蓖麻油(以商品名cremophor rh40
®
出售)、聚氧乙烯(40)蓖麻油(以商品名etocas 40
®
出售)或聚氧乙烯(60)氢化蓖麻油(以商品名nikkol hco-60
®
出售);hlb值高于10的脂肪酸乙氧基化物,比如聚氧乙烯(8)硬脂酸酯(以商品名myrj 45
®
出售)、聚氧乙烯(30)单月桂酸酯(以商品名tagat l
®
出售)、聚氧乙烯(20)硬脂酸酯(以商品名marlosol 1820
®
出售)或聚氧乙烯(15)油酸酯(以商品名marlosol ol15
®
出售);hlb值高于10的醇乙氧基化物,比如聚氧乙烯(10)油基醚(以商品名brij 96
®
出售)、聚氧乙烯(15)油基醚(以商品名volpo 015
®
出
售)、聚氧乙烯(30)油基醚(以商品名marlowet oa30
®
出售)或聚氧乙烯(20)c12-c14脂肪醚(以商品名marlowet ima20
®
出售);hlb值高于10的聚氧乙烯-聚氧丙烯共聚物和嵌段共聚物,比如hlb值=16的以商品名syperonic pe l44
®
出售的产品或hlb值=22的以商品名syperonic f127
®ꢀ
出售的产品;阴离子表面活性剂比如十二烷基硫酸钠、油酸钠或二辛基磺化琥珀酸钠或hlb值高于10的烷基酚表面活性剂,比如聚氧乙烯(9-10)壬基酚(以商品名triton n-101
®
出售)或聚氧乙烯(9)壬基酚(以商品名synperonic np9
®
出售);维生素e;d-α-生育酚聚乙二醇琥珀酸酯(tpgs)或peg 15 羟基硬脂酸酯(以商品名solutol hs15
®
出售)。
29.在一些实施方案中,至少一种亲水表面活性剂为聚乙氧基化表面活性剂。在一些实施方案中,至少一种亲水表面活性剂选自聚氧乙烯脱水山梨糖醇脂肪酸酯、聚氧乙烯烷基醚和脂肪酸的聚氧乙烯酯比如甘油和脂肪酸的聚氧乙烯酯。在一些实施方案中,脂肪酸为饱和的或不饱和的。常见的饱和脂肪酸在以上提到的表1中指出。在一些实施方案中,脂肪酸为中链脂肪酸,比如c6-c12脂肪酸(比如月桂酸、辛酸和/或癸酸)。
30.在一些实施方案中,表面活性剂中环氧乙烷基团单元的数量可为4至20之间。在一些实施方案中,至少一种亲水表面活性剂可选自聚氧乙烯(20)单油酸酯(比如商业产品吐温 80
®
)、peg 8 辛酸/癸酸甘油酯(比如商业产品labrasol
®
)、peg 6辛酸/癸酸甘油酯(比如商业产品softigel 767
®
)、聚氧乙烯(4)月桂醚(比如商业产品brij 30
®
)和它们的混合物。
31.亲水溶剂在一些实施方案中,制剂可包括至少一种无水亲水溶剂,其占制剂总重量的至多约15重量%、约10重量%、约5重量%或约1重量%的量以帮助溶解api。在一些实施方案中,制剂不含至少一种亲水溶剂。在一些实施方案中,加入至少一种亲水溶剂,比如,以溶解增稠剂。
32.在一些实施方案中,至少一种亲水溶剂可选自丙二醇、peg 400二甘醇单乙醚、三乙酸甘油酯、乙醇、甘油、异山梨醇二甲醚、n-甲基-2-吡咯烷酮、泊洛沙姆和它们的混合物。
33.化学和/或物理稳定剂在一些实施方案中,制剂可包括占制剂总重量的至多约25重量%的量的至少一种化学和/或物理稳定剂。在一些实施方案中,可加入物理稳定剂以维持在加工期间api粉末悬浮液的均匀性。如下讨论,鉴于安慰剂制剂是由溶液中的脂质赋形剂组成的单相,它是物理稳定的,但因为api粉末分散作为悬浮液,为维持均匀性,加入增稠剂。
34.化学和/或物理稳定剂可为如下的任意药物成分,其将改善制剂中不良渗透性分子化学稳定性以符合2006年6月2日的ich三方协调指导ich q3b(新药物产品中的杂质)要求当前步骤4版本,或将改善不良渗透性分子制剂物理稳定性。
35.在一些实施方案中,化学稳定剂可为亲油表面活性剂。比如,化学稳定剂可为脂肪酸甘油单酯和/或甘油二酯的醋酸、琥珀酸、乳酸、柠檬酸和/或酒石酸酯比如蒸馏乙酰化甘油单酯(以商品名myvacet 9-45
®
出售)、辛酸/癸酸二甘油琥珀酸酯(以商品名miglyol 829
®
出售)、单/双琥珀酰化甘油单酯(以商品名myverol smg
®
出售)、柠檬酸硬脂酸甘油酯(以商品名imwitor 370
®
出售)、甘油单硬脂酸/柠檬酸/乳酸酯(以商品名imwitor 375
®
出售)或甘油单酯的双乙酰酒石酸酯(以商品名cordatem t22
®
出售);由环氧乙烷和脂肪酸或脂肪酸的甘油酯反应形成的hlb值低于10的酸酯乙氧基化物,比如聚氧乙烯(4)月桂
酸(以商品名crodet 04
®
出售)、聚氧乙烯(2)硬脂酸(以商品名cithrol 2ms
®
出售);聚氧乙烯(3)硬脂酸(以商品名marlosol 183
®
出售)或甘油12 eo二油酸酯(以商品名marlowet g12do
®
出售);脂肪酸的脱水山梨糖醇酯,比如脱水山梨糖醇单月桂酸酯(以商品名span 20
®ꢀ
或 crill 1
®
出售)或脱水山梨糖醇单油酸酯(以商品名crill 4
®
出售);hlb值低于10的天然或氢化植物油甘油三酯和聚亚烷基多元醇的酯交换产品,比如聚氧乙烯化杏仁籽油(以商品名labrafil m1944cs
®
出售)、聚氧乙烯化玉米油(以商品名labrafil m2125cs
®
出售)或聚氧乙烯化氢化油(以商品名gelucire 37/06
®
出售);或hlb值低于10的醇乙氧基化物,比如聚氧乙烯化(3)油基醚(以商品名volpo n3
®
出售)、聚氧乙烯化(2)油基醚(以商品名brij 93
®
出售)或聚氧乙烯化(4)月桂醇醚(以商品名marlowet la4
®
出售)。
36.在一些实施方案中,化学稳定剂可为缓冲剂,比如柠檬酸盐、磷酸盐或醋酸盐缓冲液和/或增稠剂,比如部分氢化油、氢化油或不饱和或饱和脂肪酸的单酯、聚乙烯吡咯烷酮衍生物、聚环氧乙烷。
37.在一些实施方案中,物理稳定剂为二氧化硅。在一些实施方案中,二氧化硅可为胶态二氧化硅。胶态二氧化硅也被称为气相二氧化硅、硅灰或热解硅石。这些二氧化硅以商标aerosil
®ꢀ
(evonik industries)、cab-o-sil
®ꢀ
(cabot corporation)和 wacker hdk
®ꢀ
(waccker-chemie gmbh)商业可得。
38.在一些实施方案中,制剂可包括脂质增稠剂。脂质增稠剂的例子包括但不限于,akosoft 36、geleol、gelucire、koliwax、氢化油或它们的组合。在一些实施方案中,制剂可包括脂质增稠剂,其占制剂总重量的约5-25重量%、约10-20重量%、约12-18重量%、约14-16重量%或约15重量%的量。
39.在一些实施方案中,制剂可包括聚维酮。聚维酮的例子可包括不同等级的聚维酮比如k30或k90。在一些实施方案中,制剂可包括聚维酮,其占制剂总重量的约0.5-10重量%、约1-10重量%、约2-8重量%、约4-6重量%或约5重量%的量。
40.制剂形成在一些实施方案中,制剂可为以溶液形式的液体。在一些实施方案中,制剂为一种溶液,其中不良渗透性分子(如api)作为粉末悬浮在制剂中。在一些实施方案中,制剂可为无水反相微乳液或无水反相乳液。在一些实施方案中,制剂为均一的。均一制剂可为任意的单相或多相制剂,其可用于制造符合1999年8月3日的fda工业指导andas:共混物均匀性的散装填充物制剂,和/或用于制造符合内容物均匀性测试标准(不包括质量变化评估——欧洲药典剂量单元的均匀性2.9.40,usp总章《905》及日本药典6.02剂量单元的均匀性)的可行最终药物剂型,和/或其可满足在制造过程中取出的分层样品的稳定药物物质试验结果的合规性。
41.本文公开的制剂可根据以下过程制备。制剂可为不同赋形剂的共混物。在一些实施方案中,量最小的赋形剂可最先加入,且增稠剂可在api加入前接近最后加入。在一些实施方案中,制剂为澄清溶液且api(即不良渗透性分子或其盐)作为粉末悬浮在此制剂中。api可为纯api结晶、磨粉,经微粉化、冻干、喷雾干燥或任意本领域技术人员知晓得到固体api的方法比如常压喷雾冷冻干燥。也可为api与固体成分混合以产生固体api,比如糖苷衍生物、纤维素衍生物;或吸附于另外的赋形剂,比如介孔硅石、纳米管或任意有吸附性质的材料上;或api可被复合,诸如但不限于与离子交换树脂复合。
42.本文公开的制剂可为可消化的。因此,甘油酯可由gi汁液中的胰脂肪酶脱酯化为2-甘油单酯和游离的脂肪酸。制剂可释放癸酸钠,其可充当渗透促进剂以促进制剂中负载的不良渗透性分子的吸收。在辅脂酶存在下胰脂肪酶可催化乳化油的脂肪分解(也称为水解或脱酯化)产生脂肪酸。脂肪酸生成的速率,以及由此脂肪分解速率的测量可经由连续滴定以ph稳态追踪,如在美国专利9,259,389号中所描述,其通过引用以其全文并入本文。在37.5℃+/
−
0.5℃下,以250 mg/ml剂量在蒸馏水中包含活性为每毫克干粉末约8三丁酸甘油酯单位(tbu)的胰酶提取物的胰酶溶液中,120分钟后消化的程度可使得每g本文公开的制剂释放至少约1 mmol、约1.5 mmol或约1.7 mmol的总游离脂肪酸。
43.在一些实施方案中,在cps模型中120分钟后消化的程度(及因此消化的速率)使得每g本文公开的制剂释放至少约0.2 mmol、约0.4 mmol、约0.6 mmol或约0.7 mmol的c10 游离脂肪酸(即癸酸)。
44.在一些实施方案中,本文公开的制剂为液体或半固体(即拥有高于室温的熔融温度范围),且可使用本领域技术人员熟知的药物剂型口服施用给需要其的患者。此类药物剂型可为明胶或非明胶硬壳胶囊或软凝胶胶囊。此类胶囊可包括硬明胶胶囊和软明胶胶囊和其组合(例如,软明胶胶囊在硬明胶胶囊或非明胶软和/或硬胶囊中的覆盖封装)。此制剂也可通过本领域普通技术人员熟知的技术如吸附、热熔造粒/包衣和/或通过选定的载体、稀释剂、添加剂和/或粘结剂而转化成常规固体剂型。
45.不良渗透性分子吸收的位点可在肠中。因此,将制剂和不良渗透性分子共同递送至其吸收位点和制剂消化处是有利的。在此情况下,应避免制剂在胃中的稀释。结果,在一些实施方案中,药物剂型为包含本文公开制剂的延迟释放剂型。本领域技术人员可设想各种药物递送系统以得到延迟释放剂型。各种材料可使得能得到延迟释放效果。这些材料可被用于得到基质形式(比如ca2439366中所描述)或包衣形式。使用包衣剂型可得到一些延迟释放和保护性结果。
46.可用于制造延迟释放剂型的各种类型材料如下:对肠酶比如酯酶和脂肪酶敏感的聚合物(比如salol、虫胶、脂质化合物(硬脂酸、偏甘油酯)、巴西棕榈蜡、氢化蓖麻油)或蛋白酶(比如角蛋白、谷蛋白、玉米蛋白);可溶于肠ph的聚合物(此选择在药物工业上最广泛使用)。这些聚合物可为:多糖如果胶、纤维素或淀粉衍生物。比如纤维素乙酰邻苯二甲酸酯、羟丙基甲基纤维素、乙酰半琥珀酸纤维素、淀粉和直链淀粉乙酰邻苯二甲酸酯;乙烯基衍生物(比如,聚醋酸乙烯酯、聚乙酰邻苯二甲酸乙烯酯);丙烯酸衍生物(比如eudragit l、eudragit fs30d);或马来酸共聚物。
47.延迟释放药物剂型可为由ph决定的,且因此可使用在肠ph溶解的聚合物。在一些实施方案中,延迟释放药物剂型可为肠溶包衣剂型,特别是肠溶包衣胶囊如肠溶包衣软明胶胶囊或肠溶包衣硬壳胶囊,更特别为肠溶包衣椭圆软明胶胶囊,仍更特别为肠溶包衣7.5椭圆或更小软明胶胶囊。在一些实施方案中,根据以下指明的测试,明胶胶囊的硬度为8-12 n间,特别为9.5 n。更小的剂型对于递送不良渗透性分子进入肠内可甚至更方便。尺寸为3 mm或更小的延迟释放剂型可比更大剂型更快通过幽门的入口,且然后被患者吸收后在肠内更快释放不良渗透性分子。在这种情况下施用的剂量可要求由同时吞咽的若干小剂型组成的剂型。
48.肠溶包衣软明胶胶囊制剂的制造被本领域普通技术人员熟知,比如美国9,259,
389中所描述,其通过引用以其全文并入本文。
49.最终延迟释放药物剂型可为单片或多颗粒。这指最终剂型 (硬壳胶囊、软凝胶胶囊或其他剂型) 和中间产品(丸粒、颗粒
……
)两者皆可被包衣。特别的剂型可为多颗粒形式(填充入硬壳胶囊的包衣丸粒,用于形成若干小片剂的颗粒或丸粒)以最小化个体间的变异性。可与丙烯酸类衍生物(比如eudragit l)联合的用于肠溶包衣的增塑剂的例子如下:甘油、丙二醇、山梨醇、山梨醇/脱水山梨醇共混物、邻苯二甲酸二乙酯、邻苯二甲酸二丁酯、癸二酸二丁酯、柠檬酸三乙酯、三醋精、乙酰化甘油单酯9-45、聚乙二醇。
50.治疗活性本文公开的制剂可与包含于其中的不良渗透性分子或其盐有相同的治疗活性。因此,本公开也涉及用作药物的本文公开的肠溶药物剂型。
51.本文使用的术语“治疗有效量”可指对于治疗、改善或预防目标疾病状况,或展示可检测的治疗或预防效果所需的试剂量。通常,治疗有效剂量可基于用于在人体肠胃外施用产品可得的数据估测。
52.本文公开的化合物的有效剂量可由常规方法确定。对于任意特定患者所需要的具体剂量水平将取决于许多因素;包括被治疗状况的严重性、患者的总体健康(即年龄、重量和饮食)、患者的性别、施用的时间和频率以及对治疗的耐受/反应。总体上,然而,日剂量(不管作为单次剂量还是分次剂量施用)将在每天1至1000 mg的范围内,且最通常为每天5至200 mg。或者,可按照单位体重施用剂量,在此情况下,典型剂量将在0.01μg/kg至50 mg/kg间,尤其是在10μg/kg至10 mg/kg间、在50μg/kg至2 mg/kg间。
53.实施例1本文公开的制剂的示例组成可在以下表2中找到。
54.表2在以上制剂中api为5个氨基酸的肽,分子量约为700 g/mol。在以上制剂中,miglyol、capmul和柠檬酸三乙酯为渗透促进剂,吐温和 kolliphor el为帮助消化动力学且因而改善miglyol 和capmul的效果的表面活性剂。水和peg 400和丙二醇溶解api但对渗透性没有活性。设计制剂以作用于分子相对于肠膜的不良渗透性。胃肠道的化学和物理不稳定性和由于在胃中的酸性环境而失去活性可通过包衣解决。
55.按us 9,259,389中所描述地制备制剂1-4 (f1-f4),且作为比较品用来看通过本
文公开的发明提供的生物利用度的提高:制剂1制备:首先将api的量溶解于水中,然后加入吐温 80。搅拌得到的混合物以得到均一的溶液。然后将有规定比例的miglyol 812n 和 capmul mcm的溶液(参考表2)加入之前的混合物中。最终乳液在室温下搅拌直至得到均一的混合物(没有相分离、api完全溶解)。此制剂应当用二氧化硅来稳定。
56.制剂2制备:此制剂包含发明制剂。将有选定比例的capmul mcm 和 miglyol 812n在室温下混合在一起。然后将有规定量的吐温 80加入到溶液中。得到的混合物在室温搅拌下均一化。最后加入api量且搅拌最终混合物直至有均一的悬浮液(没有相分离、api很好地分散入填充物中)。
57.制剂3制备:此制剂为有相当低的消化性但有代替的渗透促进剂(柠檬酸三乙酯)的api的另一种溶液。首先将api的量溶解于peg400 和丙二醇的溶液中,然后加入柠檬酸三乙酯和kolliphor el。最后加入capmul mcm。得到的混合物在室温下搅拌以得到均一的溶液(没有相分离、api完全溶解)。
58.制剂4制备:将有选定比例的capmul mcm 和 miglyol 812n在室温下混合在一起。然后将有规定量的吐温 80 和柠檬酸三乙酯按顺序加入到溶液中。得到的混合物在室温搅拌下均一化。最后加入api量且搅拌最终混合物直至得到均一的悬浮液(没有相分离、api很好地分散入填充物中)。
59.用于api递送的其他示例性媒介物可包括:crodamol gmcc-ss/ 柠檬酸三乙酯/ kolliphor ei/ peg 400/ 丙二醇、miglyol 812n/ crodamol gmcc-ss/ 吐温 80、miglyol 812n/ crodamol gmcc-ss/ 柠檬酸三乙酯/ 吐温 80、miglyol 812n/ crodamol gmcc-ss/ 吐温 80且加水。
60.本文公开制剂的消化性关于可消化成分(miglyol 812n 和 capmul mcm)比例,超过85%,则制剂1(反相乳液)和2(api于悬浮液中)高度可消化。在消化30分钟后,制剂1每克制剂释放2.3 mmol的脂肪酸,制剂2每克制剂释放2.1 mmol的脂肪酸。3小时后,由制剂1和2释放的脂肪酸的最大量为每克制剂约2.8 mmol,此释放量对于所有4种制剂为可能的最大释放。在此两种制剂中超过75%的脂肪酸在少于30 分钟内释放。没有甘油三酯(miglyol 812n)的制剂3(api于溶液中)释放最低量的脂肪酸:在消化3小时后每克制剂0.6 mmol脂肪酸。在消化30 min后,每克制剂只释放了0.3 (50%)mmol的脂肪酸。相比于其他三者,由于可消化成分的水平为约70%,制剂4释放中间总量的脂肪酸(消化3小时后,每克制剂2.0 mmol的脂肪酸)。在30 分钟后释放1.7 mmol的脂肪酸,相当于在30 分钟内释放约85%。
61.以下表3示出了约700 da的五氨基酸肽的生物利用度。此肽对酶促降解不敏感,且在施用给犬后被包含于本文公开的制剂中。
62.表3制剂1234平均auc(n=6)1769459321*1878632061标准差7317102961554714439f(%)11371220*n=5。
63.使用非幼稚(non-naive)雄性比格犬(6.5-10 kg)进行了在犬中的十二指肠内施用后的药代动力学研究,以确定当在根据本发明的制剂中递送时不良渗透性分子的生物利用度。为这样做,填充物制剂通过麻醉下内窥镜的手段施用。
64.使用肌肉内注射0.03 ml/kg rompun接着肌肉内注射0.1 ml/kg zoletil 100
®
或任意相似药物,将动物麻醉。
65.使用装配有穿过内窥镜中央管的导管的塑料注射器,将测试制剂十二指肠内递送(在幽门括约肌后至少4 cm),而动物在内窥镜检测期间被安置为左侧卧。不良渗透性分子要施用的剂量按每只犬施用当天记录的体重调整,使得每只犬接收每千克动物体重相同的剂量。
66.在每次施用之前和每只动物间,用5 ml nacl 0.9%和至少20 ml空气漂洗导管。在各个时间点(通常施用前;施用后0.25、0.5、1、2、3、4、6、8、12小时),从未麻醉动物的隐静脉和头静脉收集1 ml血液样品进入柠檬酸钠管中。在样品离心(10分钟、3000 g、+4℃)之后收集血浆,且在-20℃储存直至分析。
67.静脉内施用后药代动力学研究:在静脉内注射后研究了所研究的不良渗透性分子的药代动力学,以计算其在口服或十二直肠内施用后的药代动力学参数和生物利用度。
68.犬在每次静脉内施用前被禁食持续14小时且在施用后6小时喂食(在动力学测量期间)。对于静脉内施用,使用塑料注射器将不良渗透性分子作为单次推注注射施用给犬进入外周静脉(隐静脉或头静脉)。
69.不良渗透性分子要施用的剂量按每只犬施用当天记录的体重调整,使得每只犬接收每千克动物体重相同的剂量。在各个时间点(通常施用前;施用后0.083、0.166、0.25、0.5、1、2、3、4、6、8、12小时),从未麻醉动物的隐静脉和头静脉收集1 ml血液样品进入柠檬酸钠管中。血浆样品按以上详述的制备(离心且在-20℃储存直至进一步分析)。
70.实施例2本文公开的制剂的示例组成可在以下表4中找到。
71.表4api为抗体模拟物。制剂f5等同于安慰剂制剂f2。制剂f6-f8用于测试药物载量的增加。
72.本文公开制剂的消化性:在安慰剂制剂(f5)中,游离脂肪酸的释放快速:超过85%的制剂可消化部分在少于30分钟内消化,释放已知增加通过肠膜的渗透性的游离脂肪酸(主要为c8和c10脂肪酸)。
73.制剂制造:
4-基)哌嗪-1-基)-1-氧代丙-2-基)-4-(2-氧代-1,2-二氢喹啉-3-基)哌啶-1-甲酰胺。bhv-3500见述于例如2003年12月18日公布的wo 03/104236和2013年7月9日颁发的us 8,481,546中,其通过引用全文并入本文。bhv-3500有不良渗透性并因此被选为本研究的对象。bhv-3500-d8 为bhv-3500的八氘化类似物,具有下式ii:研究在犬中单次口服bhv-3500的胶囊后bhv-3500的药代动力学(pk)。
79.材料和方法1. 实验设计和管理对于每一组,向三只雌性比格犬用bhv-3500给药一次,如表6中所示:表6. 实验设计2. 血液收集在每次给药后,于六个时间点(给药前;给药后15、30和60分钟及2和4小时)从每只犬获得血液样品(来自颈静脉,大约3ml)以测定bhv-3500的血浆水平。使用edta作为抗凝剂。血浆样品在大约-70℃下冷冻直至分析。
80.3. 参考和内部标准物及血浆样品制备提供bhv-3500和bhv-3500-d8的参考标准物并储存于室温下。这些标准物无需进一步纯化即用于制备校准标准物和质量控制(qc)样品以测定在本研究期间收集的血浆样品中的bhv-3500浓度。
81.为了测定血浆中的bhv-3500,将来自每个样品的50
ꢀµ
l等分试样转移到96孔板的适当孔中,向其中加入10
ꢀµ
l 50%的乙腈(acn)/水溶液,然后加入250
ꢀµ
l内部标准溶液(10 ng/ml的bhv-3500-d8/acn溶液)。在密封板并涡旋大约5分钟后,将板于4
±
4℃下以4,000 rpm离心10分钟。将所得上清液的一部分(100
ꢀµ
l)转移到另一个96孔板的适当孔(含有300
ꢀµ
l 0.15%的甲酸/水溶液)中。在仪器分析之前密封该板并混合其内容物。
82.新鲜制备的bhv-3500标准曲线和qc样品与研究样品一道分析。通过将10
ꢀµ
l bhv-3500储备溶液加到50
ꢀµ
l空白犬血浆中来制备仪器校准物。空白犬血浆来源于bioivt (hicksville, ny)并于-20℃下冷冻保存。标称校准物浓度在2.00至200ng/ml的范围内。以6.00、50.0和150 ng/ml的浓度制备qc样品。按照上述提取程序处理校准物和qc样品以便分析。
83.4. 分析设备和条件在表7中详述的lc-ms/ms仪器条件下分析校准物、qc和研究样品。
84.表7. 仪器操作条件
从分析物与内部标准物峰面积之比相对分析物浓度的线性回归(1/x2的加权因子)计算校准曲线。使用峰面积比和校准曲线的回归参数确定样品中的分析物浓度。
85.5. 药代动力学使用用于血管外施用的非房室模型和phoenix winnonlin软件(版本8.1;certara (新泽西州普林斯顿))分析在预定(标称)的采样时间的个体动物血浆bhv-3500浓度数据。
86.在数据允许时,通过对末期的数据点的对数线性回归(使用phoenix winnonlin的最佳拟合λz计算方法选项)来计算消除速率常数值(λz);血浆消除半衰期(t1/2)作为ln(2)/λz计算。通过线性向上/对数向下梯形规则计算从零时间值到4小时时间点浓度的血浆
浓度-时间曲线下面积值(auc0-4hr)。
87.使用标称剂量水平进行pk分析。评价了下面列出的pk参数(如果适用且在数据允许时):
•ꢀ
消除半衰期(t
1/2
)
•ꢀ
最大血浆浓度的出现时间(t
max
)
•ꢀ
最大血浆浓度(c
max
)
•ꢀ
血浆浓度-时间曲线下面积[0至4小时时间点;auc
0-4hr
]pk缩写和量度单位在表8中呈现。
[0088]
表8. pk参数定义和缩写参数单位定义rsqn/a拟合末期的线的相关性t
1/2
hr消除半衰期,由ln(2)/λz确定t
max
hr最大血浆浓度的出现时间c
max
ng/ml观察到的最大血浆药物浓度auc
0-4hr
hr*ng/ml零时间至4小时时间点的血浆浓度-时间曲线下面积结果bhv-3500浓度测定结果在表9中呈现,并以图形方式示于图1和2中。
[0089]
表9. 犬血浆中的bhv-3500浓度*下一页上继续*表9 (续). 犬血浆中的bhv-3500浓度
pk参数在表10中呈现。
[0090]
表10. pk参数分析结果:bhv-3500
*下一页上继续*表10 (续). pk参数分析结果:bhv-3500
总结当bhv-3500在无媒介物的情况下作为口服胶囊以20 mg施用于犬时(组1,无媒介物),血浆水平在所有时间点均低于定量限(bql)。只有当20 mg剂量与媒介物6和ddm组合递送(分别为组2和组3)时,血浆中的bhv-3500才可测量(参见图1)。在此20 mg剂量下,对于组2和组3,血浆bhv-3500在最早的15分钟时间点时为bql,最高浓度出现在给药后2小时,平均暴露量分别为14.2和18.9 ng/ml,这分别超过bhv-3500对人cgrp受体的亲和力(k
i = 0.023 nm)的956倍(22 nm)和1,282倍(29.5 nm)。当bhv-3500在无媒介物的情况下作为口服胶囊以50 mg施用于犬时(组4,无媒介物),在15和30分钟时间点时血浆水平为bql,在1小时和2小时时发现可测量的水平,但在4小时时没有。当50 mg剂量与媒介物6组合递送(组5)时,1小时和2小时时的血浆水平增加到3.4倍至6.6倍,平均暴露量分别为11.5和40.6 ng/ml,这分别超过bhv-3500对人cgrp受体的亲和力(k
i = 0.023 nm)的782倍(18 nm)和2,763倍(63.3 nm) (参见图2)。并且在有媒介物6的情况下,在4小时时发现可测量的血浆水平,平均暴露量为7.97 ng/ml (不同于无媒介物的情况(组4),其中在4小时时bhv-3500的血浆
水平为bql)。当50 mg剂量与ddm组合递送(组6)时,1小时和2小时时的血浆水平与无媒介物的情况(组4)相似。在有ddm的情况下(组6),在4小时时(与其中在4小时时血浆水平为bql的无媒介物情况大不相同),在4小时时发现bhv-3500可测量的血浆水平,平均暴露量为8.67 ng/ml,这超过bhv-3500对人cgrp受体的亲和力(k
i = 0.023 nm)的590倍(13.5 nm)。
[0091]
pk结果的汇总见表11中。
[0092]
表11.研究方案描述研究标题:bhv-3500在犬中的单次剂量口服胶囊研究。
[0093]
研究目的:确定犬中单次口服和舌下给药后bhv-3500胶囊制剂的药代动力学。
[0094]
研究持续时间:3周。
[0095]
试验物品制剂:标识。试验物品标识为bhv-3500。试验物品将作为胶囊供给。
[0096]
对人员的危害。遵循用于操作危险或潜在危险化学品的例行安全程序,以确保操作试验物品的人员的健康和安全。
[0097]
试验物品表征。提供验证试验物品的身份或纯度的分析证书(或其他适宜的文
件)。
[0098]
剂量制备和分析。不对给药制剂进行分析。
[0099]
储存。将bhv-3500胶囊储存于室温下。
[0100]
样品处置和保留。记录所分发的所有量的试验制品。对于该持续时间的研究,不需要保留样品。
[0101]
选择试验物品的剂量的基础。试验物品剂量水平是在先前对试验物品的pk研究的基础上选择的。
[0102]
施用途径。试验物品将口服(胶囊)施用,因为这是人类的预期施用途径之一,以及舌下施用。
[0103]
试验物品的处置。研究完成后,返回和丢弃任何剩余的试验物品。
[0104]
实验设计:参见上表6。
[0105]
试验系统:试验动物。自ridglan farms (威斯康星州mount horeb)获得三(3)或3只雌性比格犬用于本研究。所有动物均由供应商针对犬瘟热、2型腺病毒、副流感、博德特氏菌、狂犬病、乳头瘤病毒和细小病毒进行免疫接种。开始给药时,犬大约一岁龄,重大约8至12 kg。对于所有的试验物品施用,将使用相同的3只动物。
[0106]
正当理由。犬是用于非临床毒性研究的标准物种,被美国食品药品管理局认可为用于药物试剂的药代动力学安全性评估的大型动物(非啮齿动物)模型系统。
[0107]
动物数量的正当理由。所用动物的数量为获得有意义的数据所必需的最少数量。据发起人和研究负责人所知,本研究的进行不会导致关于物种、试验物品、剂量、途径和施用持续时间的现有数据的不必要重复。
[0108]
圈养。将犬单独圈养在配备自动喂水系统的围栏中。围栏每天清洁。犬根据美国农业部福利标准(第9篇,联邦法规,第3部分,1991年修订版)和“实验动物的护理和使用指南”(国家研究委员会,2011年)中规定的标准进行圈养。
[0109]
食物。经认证的犬类饮食#2021c (harlan teklad, (威斯康星州麦迪逊))。使每只犬每天可获得大约400 g食物,持续至少2小时。对每批次饮食进行污染物分析,以确保不存在预期会干扰本研究的进行或目的的浓度的污染物。来自研究中要使用的饮食批次的分析数据将保留在测试机构的文件中。给药前对犬禁食。给药后大约一小时提供食物。
[0110]
水。经由自动喂水系统向所有犬只随意提供芝加哥市粗过滤水。定期分析水的细菌污染和化学组成(例如,电解质、金属等)。水分析记录保留在测试机构的文件中。已知水中不存在预计会干扰研究的污染物。
[0111]
动物标识。每只犬都通过右耳或左耳中的usda纹身编号进行标识。在研究中,每只犬也被分配一个唯一的编号。所有围栏通过项目编号、动物编号和性别进行标识。笼卡根据组进行颜色编码。
[0112]
环境控制。每天手动记录动物房内的温度和相对湿度。使用12小时的明/暗循环(由自动计时器维持)。将动物房分别保持在大约20℃至25℃的温度和30%-70%的相对湿度范围内。
[0113]
方法:
隔离。为本研究购买的动物在施用试验物品之前被隔离至少两周。在整个隔离期间,至少每天观察一次动物的死亡或垂死迹象。
[0114]
随机化。在从隔离中释放动物后,将动物随机分组。在随机化之前,每只犬都会接受详细的临床观察以确保其适合作为试验动物。
[0115]
施用。组1至组2中的动物以20 mg/犬接受bhv-3500的单次口服胶囊施用。组4至组6中的动物以50 mg/犬的剂量接受bhv-3500的单次口服(胶囊)施用。在下一组被给药之前,每组之后将有至少48小时的清除期。
[0116]
垂死/死亡观察。在开始给药之前,至少每天观察一次动物的死亡或垂死迹象。在开始给药后并然后在整个剩余的观察期内,每天至少观察两次所有存活的研究动物的死亡或垂死迹象,并评估它们的总体健康状况。记录任何异常的临床体征。垂死/死亡检查相隔最少四小时。
[0117]
垂死的动物。在垂死/死亡观察期间,经主治兽医和研究负责人同意,任何被判断为不大可能存活到下一个预定观察期的动物将被从研究中移除、称重、安乐死和验尸。这些动物将在研究笔记本中记录为临终安乐死。死亡的动物被立即移走进行验尸并将死亡记录在研究笔记本中。
[0118]
受伤或患病的动物。根据标准兽医实践对受试动物的任何疾病或损伤进行治疗。在研究笔记本中完整记录任何受影响动物的情况和处置。任何对其他研究构成潜在感染威胁的动物将被隔离。
[0119]
临床观察。每次给药施用后大约1小时进行临床观察。
[0120]
体重管理。在每次给药前对动物称重。
[0121]
食物消耗测量。本研究中不测量个体动物的食物消耗。
[0122]
血浆药物水平。于六个时间点(给药前;每次给药后15、30和60分钟、2和4小时)从每只犬获得血液样品(从颈静脉收集大约3ml)以测定bhv-3500的血浆水平。使用edta作为抗凝剂。将血浆样品冷冻在大约-70℃下直至在测试中心分析bhv-3500的浓度。药代动力学模型包括auc、t
1/2
、t
max
和c
max
。
[0123]
事后。这是一项非终结研究。在最后一次血液收集后,犬只将被返回进行隔离。
[0124]
数据笔记本。测试中心生成的所有原始纸质数据将保存在活页笔记本中。要保存在活页笔记本中的纸质数据包括但不一定限于以下内容:
•ꢀ
原始的签署方案及任何修正和/或偏离;
•ꢀ
动物接收记录;
•ꢀ
动物护理记录;
•ꢀ
试验物品数据;
•ꢀ
血液收集数据;
•ꢀ
tk数据使用toxdata
®
以电子方式捕获的数据(例如,剂量施用、每日垂死/死亡和环境数据、临床观察、体重等)将保存在计算机系统的数据库内;toxdata
®
.htm文件的电子副本也将备份到cd-rom上并且该盘将与原始数据一起保存。
[0125]
设计的变更。方案的变更可能随研究的进展以方案修正的形式进行。未经发起人明确书面同意,不得对方案作任何更改。
[0126]
监管标准和合规性。由于本研究的试点性质,本研究将不依从美国fda制定的良好实验室规范(glp)规定(美国联邦法规第21篇第38部分)进行。本研究将按照测试中心标准操作程序进行。
[0127]
报告。准备一份报告草稿版并提交给发起人审查。报告中的信息包括但不一定限于以下内容:
•ꢀ
已批准方案的副本,包括任何修正和/或偏离
•ꢀ
所用动物的物种和品系
•ꢀ
临床观察数据
•ꢀ
体重数据
•ꢀ
血浆药物水平数据
•ꢀ
药代动力学数据在发起人审查报告草稿之后,向发起人提交最终报告。
[0128]
数据保留。作为本研究的结果产生的所有原始数据和研究的最终报告的副本将从研究完成之日起在测试中心存档一年的时间。发起人将负责与继续将档案材料存放在测试中心档案中或将这些材料运送到另一存放机构相关的所有费用。测试中心qau将保存对所有档案材料的处置的完整记录。
[0129]
人员。参与执行本研究的所有测试中心人员的简历都在测试中心存档。
[0130]
方案批准。本方案与发起人的明确文件相符。
[0131]
定义除非另外定义,否则本文使用的所有技术术语、符号和其他技术和科学术语或专业术语意图与所要求保护的主题所属领域的普通技术人员通常理解的有相同含义。在一些情况下,为阐明和/或即时参考,本文定义了有通常理解含义的术语,且本文包含的此类定义不应必然地解释为代表相比本领域通常理解的有显著差异。
[0132]
本文提及“约”数值或参数包括(和描述)针对此值或参数本身的变动。比如,提及“约x”的描述包括“x”的描述。另外,提及词组“少于”、“大于”、“至多”、“至少”、“少于或等于”、“大于或等于”或其他相似词组的描述后面有一串数值或参数,指应用该词组至该串数值或参数中的每个数值或参数。比如,制剂有至多约10重量%、约15重量%或约20重量%的组分的陈述,意在指制剂有至多约10重量%、至多约15重量%或至多约20重量%的组分。
[0133]
如本文所使用的,单数形式“一”、“一个”、“该”意图也包括复数形式,除非上下文另外清晰指示。也要理解本文使用的术语“和/或”指且包括一个或更多个相关联的列出条目的任意和所有可能的组合。进一步要理解当在本文使用术语“包括”、“包括了”、“包含”和/或“包含了”时,指明所述特征、整数、步骤、操作、元素、组分和/或单元的存在,但不排除一个或更多个其他特征、整数、步骤、操作、元素、组分、单元和/或其组的存在或加入。
[0134]
本技术在文本中公开了若干数值范围。尽管在说明书中没有逐字陈述精确范围限制,但公开的数值范围固有地支持在公开的数值范围内、包括端点的任意范围或数值,因为本公开可在整个公开的数值范围实践。
[0135]
提出以上描述以使本领域技术人员能够制造和使用本发明,且其提供于特定应用和其要求的语境中。对优选实施方案的各种修改将对本领域技术人员容易显而易见,且本文定义的一般原理可应用于其他实施方案和应用,而不偏离本公开的精神和范围。因此,本
公开不意图受限于所展示的实施方案,而要符合与本文公开的原理和特征相一致的最广的范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1