一种含有难溶性药物的浓缩液以及由其制备的乳剂的制作方法
1.本发明属于药物制剂领域。具体而言,本发明涉及一种含有难溶性药物的浓缩液及其制备方法,还涉及由所述浓缩液制备得到的乳剂。
背景技术:
2.难溶性药物给药、特别是难溶性药物的注射给药一直是药物制剂领域研究的难点和热点之一。主要原因是很多难溶性药物由于溶解度问题不能制备成注射剂,而注射剂在临床上又具有不可替代的作用,因此,增加难溶性药物的溶解度,进而实现难溶性药物的注射给药,已经成为药剂学领域最活跃的研究方向之一。
3.半个多世纪来,增加难溶性药物溶解性的策略层出不穷、不胜枚举,包括制成水包油型载药乳剂、脂质体、胶束、环糊精包合物、纳米混悬剂等,但每种方法都有其优势和局限性。
4.载药乳剂是最常见的且产业化相对成熟的制剂技术,常被用于难溶性药物注射给药的增溶,且能够获得较高的载药量。目前以乳剂形式上市的注射用难溶性药物有地西泮、联苯乙酸乙酯、氟比洛芬酯、前列地尔、地塞米松棕榈酸酯、维生素k2和丙泊酚等。虽然载药乳剂可以实现难溶性药物注射给药,但是也存在多种不足。大多数能够制成乳剂的药物通常是具有高logp值的液体或低熔点固体,而熔点高于200℃的难溶性药物不适合制成乳剂。现有载药乳剂对制剂处方、工艺、设备、配液系统均有极高要求,只有少数厂家可以掌握相关技术,并且成本高。现有市售载药乳剂的物理、化学及微生物稳定性均存在缺陷,主要表现是:分层、药物含量下降、杂质增加及容易染菌;很多乳剂不耐受高温灭菌,特别是大容量的载药乳剂注射液;现有载药乳剂注射液的储存和运输均需要冷链,成本高;现有市售载药乳剂均不耐受冻融,说明书上均明确标注“制剂不能冷冻,冷冻后应该舍弃”,为运输带来困难。难溶性药物的载药乳剂至少要保证乳剂体系12个月以上的物理及化学稳定性,这对很多难溶性药物而言是难以实现的。
5.脂质体技术也被用于难溶性药物增溶,以实现其注射给药。脂质体是指将药物包封于磷脂双分子层内而形成的微型泡囊,难溶性药物被溶解在脂质体双分子层中。以脂质体形式上市的注射用难溶性药物有紫杉醇、柔红霉素、阿霉素、两性霉素b和阿糖胞苷。脂质体作为一种难溶性药物注射给药的增溶技术,存在其局限性,例如,大规模制造的成本高,工艺复杂;另一缺点是载药量低。此外,脂质体的稳定性较差,经常需要冻干。
6.胶束制剂也是目前常见的一种增加难溶性药物溶解度、实现注射给药的方式。临床上被广泛应用的难溶性抗肿瘤药,如紫杉醇、多西他赛、卡巴他赛,都是通过聚氧乙烯蓖麻油(例如kolliphor elp)、吐温80等表面活性剂形成胶束来增溶的。但是,胶束溶液在进入血液循环后易被血液稀释。同时,由于表面活性剂用量较大,易引起一些不良反应,如溶血现象、过敏反应等,这些问题成为制约胶束作为难溶性药物注射给药增溶技术的应用瓶颈。
7.环糊精包合也是难溶性药物注射给药增溶技术的一种。目前已有多种利用环糊精
包合技术制备的注射给药产品上市,如前列地尔、伊曲康唑、伏立康唑、泊沙康唑、丝裂霉素、齐拉西酮和瑞德西韦等。但对部分难溶性药物而言,环糊精包合物载药量较低;另外,环糊精的种类较少,还具有明显的肾脏毒性,这为难溶性药物环糊精包合制剂的开发带来挑战。
8.纳米混悬剂是指药物粒子以纳米级粒径分散在含有稳定剂的水溶液中的剂型。纳米混悬剂可制成高剂量的难溶性药物制剂。该方法特别适合在水中溶解度极低或在水和油中都不溶解的难溶性药物。纳米混悬剂不需要任何辅料作为增溶剂,因此能克服其它给药系统载药量低、因加入载体而增加不良反应等缺点。采用纳米混悬技术制造的上市注射给药产品有阿扎胞苷、倍他米松、可的松醋酸酯、曲安奈德、地塞米松、甲基强的松龙、醋酸甲羟孕酮、氢化可的松等。但是纳米混悬技术存在制备工艺复杂、成本高、制剂粒径控制困难、长期储存粒径增大等不足。
9.目前仍然需要具有良好稳定性的含有难溶性药物的组合物、特别是注射用组合物。
10.基于上述背景,为了寻找含有难溶性药物的稳定组合物,发明人以塞来昔布等多种药物作为难溶性模型药物,通过各种技术,研究了不同类别的含有难溶性药物的组合物。例如,发明人制备了各种含有塞来昔布的液体制剂,如水包油乳剂、脂质体、纳米长循环混悬液、胶束等。但是,这些制剂均存在各自的问题,例如,水包油乳剂本身是一个非均相体系,在储存过程中发生分层、药物析出等问题;另外,载药乳剂的制备需要高压均质机、制剂处方、工艺、设备、配液系统均有极高要求,成本太高;而且,研究显示,塞来昔布水包油型乳剂的物理、化学及微生物稳定性均存在问题。采用聚乳酸-乙醇酸共聚物(poly(lactic-co-glycolic acid,plga)制备的纳米长循环混悬液不满足静脉注射的要求,只可能用于肌肉注射或皮下注射,限制了急救患者的使用等。
11.最后,发明人经过大量的实验研究,惊喜地发现,采用特定的复合乳化剂、油以及助乳化剂可以将临床适用剂量的难溶性药物制备成稳定的组合物,所述组合物能实现对难溶性药物的良好溶解,为均一透明的油溶液,并且制备工艺极其简单。本发明的组合物是一种浓缩液,其可在临用前用水性溶媒稀释成乳剂,所形成的乳剂直接用于对患者进行给药,用药方便,具有优良的稳定性。所述溶媒可以是适合注射的水性溶媒(例如,注射用水、5%葡萄糖注射液、0.9%氯化钠注射液等)或适合口服施用的水性溶媒(例如,纯净水、稀乙醇等)。而且,由本发明的组合物制备得到的乳剂完全满足静脉内注射的要求,可通过静脉内注射给药。
12.在中国专利申请cn108348451a中,制备了一种丙泊酚微乳剂,其是通过稀释丙泊酚浓缩物获得的,所述浓缩物包含丙泊酚、至少一种表面活性剂、至少一种溶剂、至少一种助表面活性剂和至少一种共溶剂,并且所述浓缩物大体上不含水。更具体地,在中国专利申请cn108348451a的实施例1中,制备了丙泊酚浓缩物b6,其包含丙泊酚、solutol hs-15、丙二醇、mct、peg400、乙醇、卵磷脂。与该浓缩物b6相比,本技术的浓缩物所包含的自乳化载体种类更少、制备工艺更简单,适用于更多种类的难溶性药物,并且当包含丙泊酚时导致的副作用更少。
13.因此,本发明的组合物不仅具有优良的稳定性,而且包含更少种类的自乳化载体(从而降低了载体所带来的在安全性方面的风险)、制备工艺更简单、适用于更多种类的难
溶性药物。由本发明的组合物制备得到的乳剂可顺利实现难溶性药物的给药、特别是注射给药,满足了目前未被满足的临床需求。
14.发明简述
15.本发明的目的在于提供一种含有难溶性药物的浓缩液,并且提供一种制备所述浓缩液的简单、环保、易产业化的方法。此外,本发明的目的还在于提供一种乳剂,其可顺利实现难溶性药物的给药、特别是注射给药。
16.在第一个方面,本发明提供了一种含有难溶性药物的浓缩液,其特征在于,所述浓缩液包含难溶性药物和自乳化载体,所述自乳化载体由以下物质组成:
17.(1)复合乳化剂,其由磷脂与非磷脂乳化剂组成;
18.(2)油,其是中链甘油三酸酯;和
19.(3)助乳化剂,其是无水乙醇。
20.所述的磷脂优选选自大豆磷脂、蛋黄卵磷脂及它们的混合物,更优选为蛋黄卵磷脂。
21.所述的非磷脂乳化剂优选选自聚氧乙烯蓖麻油(例如聚氧乙烯35蓖麻油、纯的聚氧乙烯35蓖麻油)、聚氧乙烯氢化蓖麻油(例如,聚氧乙烯40氢化蓖麻油、聚氧乙烯60氢化蓖麻油)、15-羟基硬脂酸聚乙二醇酯、维生素e聚乙二醇1000琥珀酸酯(tpgs)、聚山梨酯(例如聚山梨酯20、21、40、60、61、65、80、81、85、120,特别是聚山梨酯80)及它们的混合物。更优选地,所述的非磷脂乳化剂选自聚氧乙烯40氢化蓖麻油、聚氧乙烯35蓖麻油、纯的聚氧乙烯35蓖麻油、15-羟基硬脂酸聚乙二醇酯、聚山梨酯80及它们的混合物。
22.在该方面的一个实施方案中,在本发明所述的含有难溶性药物的浓缩液中,当将难溶性药物、复合乳化剂、油、助乳化剂的重量总计为100重量%时,所述难溶性药物的重量百分比为0.01%~20%,优选0.1%~15%,更优选0.1%~12%,例如0.1%、0.5%、1%、1.5%、2%、5%、6%、10%、12%;所述磷脂的重量百分比为0.5%~10%,优选为1%~5%,例如1%、2%、3%、4%、5%;所述非磷脂乳化剂的重量百分比为30%~70%,优选为30%~60%,更优选30%~50%,例如30%、31%、32%、33%、39%、40%、43%、44%、46%、47%、48%、49%、50%;所述中链甘油三酸酯的重量百分比为20%~50%,优选20%~40%,更优选23%~40%,例如20%、23%、24%、25%、28%、29%、30%、32%、33%、35%、37%、40%;余量为无水乙醇。
23.上文所述的含有难溶性药物的浓缩液可以仅由以下组分组成:难溶性药物;复合乳化剂,其由磷脂与非磷脂乳化剂组成;油,其是中链甘油三酸酯;和助乳化剂,其是无水乙醇。
24.或者,上文所述的含有难溶性药物的浓缩液还可以含有ph调节剂和/或抗氧化剂。所述的ph调节剂可以选自枸橼酸、枸橼酸盐(如枸橼酸钠)、马来酸、酒石酸、盐酸、氢氧化钠、醋酸、醋酸盐(如醋酸钠)、磷酸、磷酸盐(如磷酸一氢钠、磷酸二氢钠或磷酸钠)中的一种或多种。所述的抗氧化剂可以选自α-生育酚琥珀酸酯(α-tocopherol succinate)、棕榈酸抗坏血酸酯(ascorbyl palmitate)、丁基化羟基苯甲醚(butylated hydroxyanisole,bha)、丁化羟基甲苯(butylated hydroxytoluene,bht)中的一种或多种。
25.在第二个方面,本发明提供了制备所述的含有难溶性药物的浓缩液的方法,其特征在于,所述方法包括以下步骤:将难溶性药物、磷脂、非磷脂乳化剂、作为油的中链甘油三
酸酯和作为助乳化剂的无水乙醇以任意顺序混合,搅拌均匀,过滤,分装压盖密封。
26.在第三个方面,本发明提供了一种乳剂,其是通过将上文所述的含有难溶性药物的浓缩液用水性溶媒稀释制备的,所述乳剂的平均粒径在20nm~4000nm之间,优选在20nm~1000nm之间,更优选在20nm~500nm之间,进一步优选在20nm~300nm之间。
27.在第四个方面,本发明提供了上文所述的含有难溶性药物的浓缩液在制备乳剂中的用途。所述乳剂尤其可用于静脉注射。
附图说明
28.图1显示了实施例7中用供试品(即,将本发明的浓缩液2用5%葡萄糖注射液以1g:100ml的比例稀释所获得的塞来昔布注射液)、阳性对照(即,市售的帕瑞昔布钠注射液和氟比洛芬酯注射液)、阴性对照(即,5%葡萄糖注射液)、空白制剂(将供试品中的难溶性药物替换为相同重量的无水乙醇所获得的制剂)在大鼠切口痛镇痛模型中产生的药效学实验结果。图1的纵坐标是50%缩足反应阈值(g),横坐标是时间(小时)。其中,第一条曲线(
▲
)代表供试品塞来昔布注射液的实验结果,第二条曲线(
■
)是代表阳性对照氟比洛芬酯注射液的实验结果,第三条曲线(
●
)代表阳性对照帕瑞昔布钠注射液的实验结果,第四条曲线(
○
)代表空白制剂的实验结果,第五条曲线代表阴性对照5%葡萄糖注射液的实验结果。在图1中,***表示供试品塞来昔布乳剂注射液与帕瑞昔布钠注射液相比具有显著性差异p《0.01,##表示供试品塞来昔布乳剂注射液与氟比洛芬酯注射液相比具有显著性差异p《0.05,###表示本发明的塞来昔布乳剂注射液与氟比洛芬酯注射液相比具有显著性差异p《0.01。
29.发明详述
具体实施方案
30.实施方案1.一种浓缩液,其特征在于,所述浓缩液包含难溶性药物和自乳化载体,所述自乳化载体由以下物质组成:
31.(1)复合乳化剂,其由磷脂与非磷脂乳化剂组成;
32.(2)油,其是中链甘油三酸酯;和
33.(3)助乳化剂,其是无水乙醇,
34.其中所述磷脂选自大豆磷脂、蛋黄卵磷脂及它们的混合物。
35.实施方案2.根据实施方案1所述的浓缩液,其中所述非磷脂乳化剂选自聚氧乙烯蓖麻油(例如,聚氧乙烯35蓖麻油、纯的聚氧乙烯35蓖麻油)、聚氧乙烯氢化蓖麻油(例如,聚氧乙烯40氢化蓖麻油、聚氧乙烯60氢化蓖麻油)、15-羟基硬脂酸聚乙二醇酯、维生素e聚乙二醇1000琥珀酸酯(tpgs)、聚山梨酯(例如聚山梨酯20、21、40、60、61、65、80、81、85、120,特别是聚山梨酯80)及它们的混合物。
36.实施方案3.根据实施方案2所述的浓缩液,其中所述的非磷脂乳化剂选自聚氧乙烯40氢化蓖麻油、聚氧乙烯35蓖麻油、纯的聚氧乙烯35蓖麻油、15-羟基硬脂酸聚乙二醇酯、聚山梨酯80及它们的混合物。
37.实施方案4.根据实施方案1至3中任意一项所述的浓缩液,其中当将难溶性药物、复合乳化剂、油、助乳化剂的重量总计为100重量%时,所述难溶性药物的重量百分比为
0.01%~20%,优选0.1%~15%,更优选0.1%~12%;所述磷脂的重量百分比为0.5%~10%,优选为1%~5%;所述非磷脂乳化剂的重量百分比为30%~70%,优选为30%~60%,更优选30%~50%;所述中链甘油三酸酯的重量百分比为20%~50%,优选20%~40%,更优选23%~40%;余量为无水乙醇。
38.实施方案5.根据实施方案1至4中任意一项所述的浓缩液,其中所述难溶性药物选自塞来昔布(celecoxib)、伐地昔布(valdecoxib)、依托考昔(etocoxib)、布洛芬(ibuprofen)、右旋布洛芬(dexibuprofen)、丙泊酚(propofol)、氟比洛芬酯(flurbiprofen axetil)、前列地尔(alprostadil)、丁酸氯维地平(clevidipine butyrate)、地塞米松棕榈酸酯(dexamethasone palmitate)、非洛地平(felodipine)、尼莫地平(nimodipine)、硝苯地平(nifedipine)、尼群地平(nitrendipine)、环孢素(ciclosporin)、他克莫司(tacrolimus)、左西孟旦(levosimendan)、阿德福韦酯(adefovir dipivoxil)、红霉素(erythromycin)、罗红霉素(roxithromycin)、泊沙康唑(posaconazole)、伊曲康唑(itraconazole)、伏立康唑(voriconazole)、咪康唑(miconazole)、酮康唑(ketoconazole)、黄体酮(progesterone)、辅酶q10(coenzyme q10)、氯吡格雷(clopidogrel)、紫杉醇(paclitaxel)、多西他赛(docetaxel)、卡巴他赛(cabazitaxel)、依托泊苷(etoposide)、替尼泊苷(teniposide)、羟基喜树碱(hydroxycamptothecin)、伊立替康(irinotecan)、乌苯美司(ubenimex)、顺铂(cisplatin)、卡铂(carboplatin)、卡培他滨(capecitabine)、奥沙利铂(oxaliplatin)、吉非替尼(gefitinib)、多柔比星(doxorubicin)、长春碱(vinblastine)、长春新碱(vincristine)、长春西汀(vinpocetine)、长春地辛(vindesine)、吡罗昔康(piroxicam)、螺内酯(spironolactone)、丙戊酸(valproic acid)、他莫昔芬(tamoxifen)、阿奇霉素(azithromycin)、维生素a、维生素d、维生素e、维生素k、非诺贝特(fenofibrate)、吲哚美辛(indomethacin)、瑞德西韦(remdesivir);优选地,所述难溶性药物选自塞来昔布、布洛芬、右旋布洛芬、丙泊酚、氟比洛芬酯、前列地尔、丁酸氯维地平、地塞米松棕榈酸酯、非洛地平、尼莫地平、硝苯地平、尼群地平、环孢素、他克莫司、左西孟旦、阿德福韦酯、红霉素、罗红霉素、泊沙康唑、伊曲康唑、伏立康唑、黄体酮、辅酶q10、氯吡格雷、紫杉醇、多西他赛、卡巴他赛、依托泊苷、替尼泊苷、丁酸氯维地平、瑞德西韦、卡铂、伐地昔布、阿奇霉素和依托考昔;最优选地,所述难溶性药物选自塞来昔布、紫杉醇、多西他赛、布洛芬、尼莫地平、辅酶q10、卡巴他赛、依托考昔、泊沙康唑、环孢素、氟比洛芬酯、右旋布洛芬、左西孟旦、丁酸氯维地平、氯吡格雷、瑞德西韦、他克莫司、卡铂、地塞米松棕榈酸酯、伐地昔布、阿奇霉素和丙泊酚。
39.实施方案6.根据实施方案1至5中任意一项所述的浓缩液,其还含有ph调节剂、抗氧化剂或这二者。
40.实施方案7.根据实施方案1至5中任意一项所述的浓缩液,其中
41.所述的难溶性药物是塞来昔布,所述的磷脂是蛋黄卵磷脂,所述的非磷脂乳化剂是15-羟基硬脂酸聚乙二醇酯,所述的油是中链甘油三酸酯,所述的助乳化剂是无水乙醇,并且当将难溶性药物、复合乳化剂、油、助乳化剂的重量总计为100重量%时,塞来昔布的重量百分比为5%,蛋黄卵磷脂的重量百分比为3%,15-羟基硬脂酸聚乙二醇酯的重量百分比为48%,中链甘油三酸酯重量百分比为28%,无水乙醇重量百分比为16%;
42.或者
43.所述的难溶性药物是布洛芬,所述的磷脂是蛋黄卵磷脂,所述的非磷脂乳化剂是15-羟基硬脂酸聚乙二醇酯,所述的油是中链甘油三酸酯,所述的助乳化剂是无水乙醇,所述浓缩液还包含枸橼酸,并且当将难溶性药物、复合乳化剂、油、助乳化剂的重量总计为100重量%时,布洛芬的重量百分比为12%,蛋黄卵磷脂的重量百分比为2%,15-羟基硬脂酸聚乙二醇酯的重量百分比为48%,中链甘油三酸酯的重量百分比为27%,无水乙醇的重量百分比为11%;
44.或者
45.所述的难溶性药物是多西他赛,所述的磷脂是蛋黄卵磷脂,所述的非磷脂乳化剂是15-羟基硬脂酸聚乙二醇酯,所述的油是中链甘油三酸酯,所述的助乳化剂是无水乙醇,并且当将难溶性药物、复合乳化剂、油、助乳化剂的重量总计为100重量%时,多西他赛的重量百分比为2%,蛋黄卵磷脂的重量百分比为3%,15-羟基硬脂酸聚乙二醇酯的重量百分比为48%,中链甘油三酸酯的重量百分比为28%,无水乙醇的重量百分比为19%;
46.或者
47.所述的难溶性药物是紫杉醇,所述的磷脂是蛋黄卵磷脂,所述的非磷脂乳化剂是15-羟基硬脂酸聚乙二醇酯,所述的油是中链甘油三酸酯,所述的助乳化剂是无水乙醇,所述浓缩液还包含枸橼酸,并且当将难溶性药物、复合乳化剂、油、助乳化剂的重量总计为100重量%时,紫杉醇的重量百分比为1.5%,蛋黄卵磷脂的重量百分比为3%,15-羟基硬脂酸聚乙二醇酯的重量百分比为49.1%,中链甘油三酸酯的重量百分比为29.4%,无水乙醇的重量百分比为17%。
48.实施方案8.实施方案1至7中任意一项所述的浓缩液在制备乳剂、特别是用于静脉内注射、例如用于静脉内滴注的乳剂中的用途。
49.实施方案9.根据实施方案8所述的用途,所述乳剂的平均粒径在20nm~4000nm之间,优选在20nm~1000nm之间,更优选在20nm~500nm之间,进一步优选在20nm~300nm之间。
50.实施方案10.制备实施方案1至7中任意所述的浓缩液的方法,所述方法包括以下步骤:将难溶性药物、磷脂、非磷脂乳化剂、中链甘油三酸酯和无水乙醇以任意顺序混合,搅拌均匀,过滤,分装压盖密封。
51.实施方案11.一种乳剂,其是通过将实施方案1至7中任意一项所述的浓缩液用水性溶媒稀释获得的。
52.实施方案12.根据实施方案11所述的乳剂,其中所述水性溶媒是适合注射的水性溶媒,选自注射用水、5%葡萄糖注射液和0.9%氯化钠注射液。
53.实施方案13.根据实施方案12所述的乳剂,其用于静脉内注射,特别是静脉内滴注。
54.本发明的组合物及其制备方法
55.发明人通过对难溶性药物的基本理化性质的研究,发现可以将难溶性药物例如塞来昔布等用由磷脂和非磷脂乳化剂组成的复合乳化剂以及特定的助乳化剂和油制备成稳定的组合物,其是一种浓缩液,在临用前用水性溶媒稀释形成乳剂,例如,用适合注射的水性溶媒如注射用水、5%葡萄糖注射液或0.9%氯化钠注射液稀释形成乳剂,即可直接用于注射给药,例如静脉内注射给药,特别是静脉内滴注给药。
56.因此,在第一个方面,本发明提供了一种含有难溶性药物的浓缩液,其特征在于,该组合物包含:难溶性药物;复合乳化剂,其中所述复合乳化剂由磷脂与非磷脂乳化剂组成;油,其是中链甘油三酸酯;和助乳化剂,其是无水乙醇。
57.所述的磷脂优选选自大豆磷脂、蛋黄卵磷脂及它们的混合物,更优选为蛋黄卵磷脂。
58.所述非磷脂乳化剂选自聚氧乙烯蓖麻油(例如,聚氧乙烯35蓖麻油、纯的聚氧乙烯35蓖麻油)、聚氧乙烯氢化蓖麻油(例如,聚氧乙烯40氢化蓖麻油、聚氧乙烯60氢化蓖麻油)、15-羟基硬脂酸聚乙二醇酯、维生素e聚乙二醇1000琥珀酸酯(tpgs)、聚山梨酯(例如聚山梨酯20、21、40、60、61、65、80、81、85、120,特别是聚山梨酯80)及它们的混合物。更优选地,所述的非磷脂乳化剂选自聚氧乙烯40氢化蓖麻油、聚氧乙烯35蓖麻油、纯的聚氧乙烯35蓖麻油、15-羟基硬脂酸聚乙二醇酯、聚山梨酯80及它们的混合物。
59.在第一个方面的一个优选的实施方案中,在本发明所述的含有难溶性药物的浓缩液中,当将难溶性药物、复合乳化剂、油、助乳化剂的重量总计为100重量%时,所述难溶性药物的重量百分比为0.01%~20%,优选0.1%~15%,更优选0.1%~12%,例如0.1%、0.5%、1%、1.5%、2%、5%、6%、10%、12%;所述磷脂的重量百分比为0.5%~10%,优选为1%~5%,例如1%、2%、3%、4%、5%;所述非磷脂乳化剂的重量百分比为30%~70%,优选为30%~60%,更优选30%~50%,例如30%、31%、32%、33%、39%、40%、43%、44%、46%、47%、48%、49%、50%;所述中链甘油三酸酯的重量百分比为20%~50%,优选20%~40%,更优选23%~40%,例如20%、23%、24%、25%、28%、29%、30%、32%、33%、35%、37%、40%;余量为无水乙醇。
60.本发明的含有难溶性药物的浓缩液是均一透明的油溶液,具有良好的物理和化学稳定性。在室温下放置6个月,一直是均一透明的油溶液,未发生分层,也未见药物析出。例如,在40
±
2℃/60
±
5%rh条件下考察了本发明的实施例4中所制备的浓缩液的化学稳定性,结果显示,在温度40℃
±
2℃、相对湿度60%
±
5%的条件下放置6个月,浓缩液中的难溶性药物的含量和有关物质均未发生明显变化,符合药品质控标准要求。
61.本发明的含有难溶性药物的浓缩液可以用水性溶媒稀释,从而形成乳剂。所述水性溶媒可以是适合注射的水性溶媒(例如,注射用水、5%葡萄糖注射液、0.9%氯化钠注射液等)或适合口服施用的水性溶媒(例如,纯净水、稀乙醇等)。在用水性溶媒稀释后,本发明的含有难溶性药物的浓缩液形成的乳剂具有的平均粒径在20nm~4000nm之间,优选在20nm~1000nm之间,更优选在20nm~500nm之间,进一步优选在20nm~300nm之间。用适合注射的水性溶媒例如5%葡萄糖注射液稀释后,本发明的浓缩液所形成的乳剂具有的平均粒径不超过4000nm,符合静脉内注射剂、甚至是静脉内滴注的注射剂的要求,因此,所述乳剂可用于皮下注射、皮内注射、腹膜内注射,也可用于静脉内注射,包括静脉内推注和静脉内滴注。
62.本发明的浓缩液不仅自身具有良好的稳定性,而且由其稀释获得的乳剂也具有良好的稳定性。例如,将本发明的浓缩液、具体是浓缩液1用5%葡萄糖注射液以1g:100ml的比例稀释后形成的乳剂于室温条件下放置24小时,未见药物析出或分层,ph值稳定在4.5~7.0之间。
63.此外,本发明的浓缩液还具有一些额外的优势。具体而言,与中国专利申请cn108348451a所公开的浓缩物相比,本发明的组合物所包含的自乳化载体种类更少、制备
工艺更简单,适用于更多种类的难溶性药物,并且副作用更少。
64.本文所用的术语“难溶性药物”是指已知的在医药领域中可以应用的药物,且相对其有效给药量而言,其在水中的溶解度较低。更具体地,本文所述的“难溶性药物”是指属于中国药典“凡例”中记载的溶解度为“微溶”(溶质1g(ml)能在溶剂100~不到1000ml中溶解)、“极微溶解”(溶质1g(ml)能在溶剂1000~不到10000ml中溶解)或“几乎不溶或不溶”(溶质1g(ml)在溶剂10000ml中不能完全溶解)的药物。
65.本文所述的难溶性药物的实例包括、但不限于:塞来昔布、伐地昔布、依托考昔、布洛芬、右旋布洛芬、丙泊酚、氟比洛芬酯、前列地尔、丁酸氯维地平、地塞米松棕榈酸酯、非洛地平、尼莫地平、硝苯地平、尼群地平、环孢素、他克莫司、左西孟旦、阿德福韦酯、红霉素、罗红霉素、泊沙康唑、伊曲康唑、伏立康唑、咪康唑、酮康唑、黄体酮、辅酶q10、氯吡格雷、紫杉醇、多西他赛、卡巴他赛、依托泊苷、替尼泊苷、羟基喜树碱、伊立替康、乌苯美司、顺铂、卡铂、卡培他滨、奥沙利铂、吉非替尼、多柔比星、长春碱、长春新碱、长春西汀、长春地辛、吡罗昔康、螺内酯、丙戊酸、他莫昔芬、阿奇霉素、维生素a、维生素d、维生素e、维生素k、非诺贝特、吲哚美辛、瑞德西韦。优选地,所述难溶性药物选自塞来昔布、布洛芬、右旋布洛芬、丙泊酚、氟比洛芬酯、前列地尔、丁酸氯维地平、地塞米松棕榈酸酯、非洛地平、尼莫地平、硝苯地平、尼群地平、环孢素、他克莫司、左西孟旦、阿德福韦酯、红霉素、罗红霉素、泊沙康唑、伊曲康唑、伏立康唑、黄体酮、辅酶q10、氯吡格雷、紫杉醇、多西他赛、卡巴他赛、依托泊苷、替尼泊苷、丁酸氯维地平、瑞德西韦、卡铂、伐地昔布、阿奇霉素和依托考昔;最优选地,所述难溶性药物选自塞来昔布、紫杉醇、多西他赛、布洛芬、尼莫地平、辅酶q10、卡巴他赛、依托考昔、泊沙康唑、环孢素、氟比洛芬酯、右旋布洛芬、左西孟旦、丁酸氯维地平、氯吡格雷、瑞德西韦、他克莫司、卡铂、地塞米松棕榈酸酯、伐地昔布、阿奇霉素和丙泊酚。
66.发明人对本发明的浓缩液中所用的油进行了考察,实验结果显示,当使用中链甘油三酸酯时获得了最佳的稳定性。如果将中链甘油三酸酯用注射用大豆油、橄榄油、鱼油、玉米油、结构油、蓖麻油、葵花籽油、棉籽油、茶油等替换,所得的组合物均出现分层,体系不均一,制剂成型性差。
67.发明人还对本发明的浓缩液中所用的乳化剂进行了筛选,磷脂乳化剂考察了大豆磷脂、蛋黄卵磷脂、氢化大豆卵磷脂;非磷脂乳化剂考察了聚氧乙烯40氢化蓖麻油(例如kolliphor rh40)、聚氧乙烯35蓖麻油(例如kolliphor el和kolliphor elp)、15-羟基硬脂酸聚乙二醇酯(例如kolliphor hs15)、维生素e聚乙二醇琥珀酸酯(tpgs)、聚山梨酯80(例如吐温80)。实验结果显示,使用单独的磷脂乳化剂或非磷脂乳化剂制备得到的组合物在稀释成乳剂后所得的乳剂稳定性均较差,在形成乳剂后2~4小时左右便会分层;只有将磷脂和非磷脂乳化剂这二者组合使用时,所得的浓缩液才稳定、均一,并且在稀释成乳剂后所得乳剂具有良好的稳定性,满足静脉注射给药制剂的所有要求。
68.发明人还对助乳化剂进行了考察,实验结果显示,当使用无水乙醇时获得了最佳的稳定性。如果将乙醇用丙二醇、甘油、peg200、peg300、peg400等替换,所得的组合物均分层,体系不均一,制剂不可成型。
69.本发明的浓缩液最优选是具有本技术的实施例4中所给出的组分和比例的那些。
70.上文所述的本发明的含有难溶性药物的浓缩液可以仅由以下组分组成:难溶性药物;复合乳化剂,其由磷脂与非磷脂乳化剂组成;油,其是中链甘油三酸酯;和助乳化剂,其
是无水乙醇。
71.或者,上文所述的本发明的含有难溶性药物的浓缩液还可以含有其它组分,例如ph调节剂和/或抗氧化剂。所述的ph调节剂可以选自枸橼酸、枸橼酸盐(如枸橼酸钠)、马来酸、酒石酸、盐酸、氢氧化钠、醋酸、醋酸盐(如醋酸钠)、磷酸、磷酸盐(如磷酸一氢钠、磷酸二氢钠或磷酸钠)中的一种或多种。所述的抗氧化剂可以选自α-生育酚琥珀酸酯、棕榈酸抗坏血酸酯、丁基化羟基苯甲醚(bha)、丁化羟基甲苯(bht)中的一种或多种。
72.在第二个方面,本发明提供了制备所述的含有难溶性药物的浓缩液的方法,其特征在于,所述方法包括以下步骤:将难溶性药物、磷脂、非磷脂乳化剂、中链甘油三酸酯和无水乙醇以任意顺序混合,搅拌均匀,过滤,分装压盖密封。
73.发明人发现,制备本发明的浓缩液的方法非常简单,各个组分的加入顺序、搅拌时间等对浓缩液的品质没有影响,只要确保难溶性药物溶解即可。
74.本发明采用极其简单的处方工艺,制备出了具有良好稳定性的含有难溶性药物的浓缩液,其经水性溶媒稀释即可获得直接用于静脉注射给药的乳剂。相比于现有技术,本发明的浓缩液配方简化,制备工艺简单,只需通过简单的混合、搅拌、过滤、封装即可获得,无需均质机、微射流仪及复杂的配液系统,一般企业都可以实现该工艺。而且,本发明的浓缩液物理、化学稳定性显著提高;储存和运输无需冷链,极大地降低了生产、运输、储存及使用的成本,为临床用药提供巨大方便。
75.在第三个方面,本发明提供了一种乳剂,其是通过将上文所述的含有难溶性药物的浓缩液用水性溶媒稀释获得的,所述乳剂的平均粒径在20nm~4000nm之间,优选在20nm~1000nm之间,更优选在20nm~500nm之间,进一步优选在20nm~300nm之间。
76.所述乳剂在24h内具有良好的稳定性。
77.所述乳剂可以施用于患者,以治疗其中所包含的难溶性药物所能治疗的疾病。例如,包含塞来昔布的乳剂可用于治疗急性疼痛和炎性疾病例如骨关节炎和类风湿性关节炎;包含紫杉醇或多西他赛的乳剂可用于治疗癌症,例如实体瘤,如乳腺癌、卵巢癌、头颈癌、肺癌(包括非小细胞肺癌和小细胞肺癌)、胰腺癌、胃癌、黑色素瘤、软组织肉瘤;包含布洛芬或右旋布洛芬的乳剂可用于治疗疼痛,例如头痛、关节痛、偏头痛、牙痛、肌肉痛、神经痛、痛经。本文所述的难溶性药物及其用途均是本领域已知的,描述这些难溶性药物的用途的现有技术文献均视为本技术的一部分。
78.在第四个方面,本发明提供了上文所述的含有难溶性药物的浓缩液在制备乳剂中的用途。所述乳剂尤其可用于静脉注射,例如静脉推注和静脉滴注。
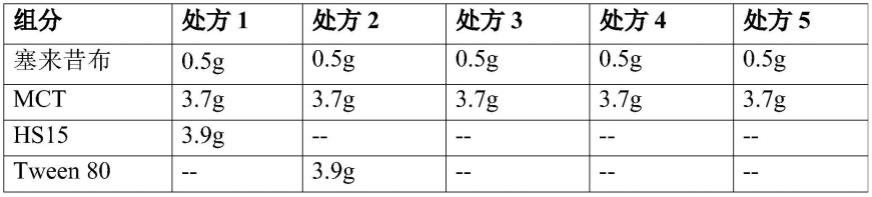
79.在本公开内容中,发明人以塞来昔布等多种药物作为难溶性药物的模型,详细考察了影响含有难溶性药物的浓缩液的形成、物理和化学稳定性、稀释后所得乳剂的物理稳定性及药效的主要因素,获得了本发明的浓缩液。该浓缩液可广泛应用于难溶性药物,实现了难溶性药物的注射给药,为临床应用提供新的治疗可能。
80.定义
81.在本技术的上下文中,“含有难溶性药物的组合物”、“含有难溶性药物的浓缩液”、“本发明的组合物”、“本发明的浓缩液”等术语可以互换使用,均指包含难溶性药物、由磷脂与非磷脂乳化剂组成的复合乳化剂、作为油的中链甘油三酸酯和作为助乳化剂的无水乙醇的组合物,上下文显示含义并非如此的除外。
82.本文所用的术语“自乳化载体”是指在将本发明的浓缩液用水性溶媒稀释时有助于形成乳剂的药学上可接受的载体。
83.本文所用的术语“由
……
组成”是指除了所列明的组份外不包含显著量的其它物质。例如,在本发明的浓缩液中,自乳化载体由复合乳化剂、油和助乳化剂组成,这意味着除了本文所定义的复合乳化剂、油和助乳化剂之外,不包含显著量的其它有助于形成乳剂的物质。
84.本文所用的术语“中链甘油三酸酯”是指从椰子坚硬干燥的内胚乳或者油椰干燥的内胚乳中提取的非挥发性植物油,是由饱和脂肪酸甘油三酸酯组成的混合物。中链甘油三酸酯可以商购获得,例如从辽宁新兴药业、德国ioi oleo gmbh等商购获得。
85.本文所用的术语“无水乙醇”是指纯度高达99.5%及以上的乙醇。
86.本文所用的术语“聚氧乙烯蓖麻油”是指由不同量环氧乙烷和蓖麻油反应得到的物质。聚氧乙烯蓖麻油的实例包括、但不限于聚氧乙烯35蓖麻油、纯的聚氧乙烯35蓖麻油。
87.本文所用的术语“聚氧乙烯35蓖麻油”是指由1mol甘油蓖麻酸酯与35mol环氧乙烷反应得到的物质,其中除了聚氧乙烯甘油三蓖麻酸酯之外,还含有少量的聚乙二醇蓖麻酸酯和游离乙二醇。聚氧乙烯35蓖麻油可以商购获得,例如以商品名kolliphor el和kolliphor elp从巴斯夫公司(basf)等商购获得。
88.本文所用的术语“纯的聚氧乙烯35蓖麻油”是指纯化的聚氧乙烯甘油三蓖麻酸酯,基本上不含聚乙二醇蓖麻酸酯和游离乙二醇。
89.本文所用的术语“聚氧乙烯氢化蓖麻油”是指由不同量环氧乙烷和氢化蓖麻油反应得到的物质。聚氧乙烯氢化蓖麻油的实例包括、但不限于聚氧乙烯40氢化蓖麻油、聚氧乙烯60氢化蓖麻油。
90.本文所用的术语“聚氧乙烯40氢化蓖麻油”是指由l mol甘油三羟基硬脂酸与40-45mol环氧乙烷反应得到的物质,其中除了聚氧乙烯甘油三羟基硬脂酸酯之外,还含有少量聚乙二醇三羟基硬脂酸、游离的聚乙二醇。聚氧乙烯40氢化蓖麻油可以商购获得,例如以商品名kolliphor rh40从巴斯夫公司(basf)等商购获得。
91.本文所用的术语“聚氧乙烯60氢化蓖麻油”是指由l mol甘油三羟基硬脂酸与60mol环氧乙烷反应得到的物质,其中除了聚氧乙烯甘油三羟基硬脂酸酯之外,还含有少量聚乙二醇三羟基硬脂酸、游离的聚乙二醇。
92.本文所用的术语“15-羟基硬脂酸聚乙二醇酯”可以商购获得,例如以商品名kolliphor hs15或solutol hs-15从巴斯夫公司(basf)或sigma-aldrich商购获得。
93.本文所用的术语“维生素e聚乙二醇琥珀酸酯(tpgs)”是一种维生素e的水溶性衍生物,由维生素e琥珀酸酯的羧基与聚乙二醇的羟基反应而成,其可以商购获得,例如以商品名tocofersolan(tpgs)从巴斯夫公司(basf)商购获得。
94.本文所用的术语“聚山梨酯”是指一系列聚氧乙烯去水山梨醇的部分脂肪酸酯,其由每摩尔山梨醇与大约20.5mol或4mol环氧乙烷的比例共聚而成。聚山梨酯的实例包括、但不限于例如聚山梨酯20、21、40、60、61、65、80、81、85、120,特别是聚山梨酯80。聚山梨酯可以商购获得,例如以商品名吐温20、吐温40、吐温80等从南京威尔药业股份有限公司商购获得。
95.本文所用的术语“约”、“大约”、“左右”表示其后所给出的数值可以上下扩展20%。
例如,“约100”表示80%至120%。
96.本文所用的术语“基本上不含”是指其后的物质的含量小于1%。
97.实施例
98.以下实施例用于对本发明进行举例说明,但不以任何方式限制所附的权利要求所定义的范围。
99.除非另有说明,否则粒径和多分散系数(pdi)是使用激光粒度仪(美国pss,型号nicompz3000)测定的,ph值测定是使用ph计(pb-10,赛多利斯)测定的,高效液相色谱法是使用岛津lc-20at进行的,所给出的温度单位是摄氏度,未说明温度的操作是在环境温度下进行的。
100.实施例中所用的缩略语的含义如表1所示。
101.表1—缩略语含义
102.缩略语含义生产商elp聚氧乙烯35蓖麻油kolliphor elbasfepc蛋黄卵磷脂epc 80lipoidspc大豆磷脂江苏曼氏生物hspc氢化大豆卵磷脂lipoidrh40聚氧乙烯40氢化蓖麻油kolliphor rh40basfhs1515-羟基硬脂酸聚乙二醇酯kolliphor hs15basftween 80吐温80南京威尔药业tpgs聚乙二醇1000维生素e琥珀酸酯basfmct中链甘油三酸酯辽宁新兴药业lct大豆油辽宁新兴药业
103.实施例1:乳化剂的筛选
104.1、处方1~5:单一乳化剂的筛选
105.表2—包含单一乳化剂的处方
[0106][0107][0108]
工艺:将处方量的各组分加入20ml的小瓶中,磁力搅拌,直至所有组分完全溶解。
[0109]
处方1-5所得的浓缩液均为均一透明的油溶液。将所得油溶液以1g:100ml的比例用5%葡萄糖注射液稀释。在室温下放置,观察形成乳剂的情况并考察所得乳剂的稳定性,
结果见表3。
[0110]
表3—用单一乳化剂制备的浓缩液稀释后形成的乳剂的稳定性
[0111][0112]
上述实验数据表明,使用hs15、tween 80、elp、tpgs或rh40作为单一乳化剂制备的浓缩液虽然都是均一透明的油溶液,并且用5%葡萄糖注射液稀释后均形成白色乳剂,但是,所形成的乳剂迅速发生分层,表明乳剂稳定性差。
[0113]
鉴于用单一乳化剂制备的任何一种浓缩液在稀释后均无法形成稳定的乳剂,未对这些乳剂的粒径进行测定。
[0114]
2、处方6~11:复合乳化剂的筛选
[0115]
表4—包含复合乳化剂的处方
[0116][0117][0118]
工艺:将处方量的各组分加入20ml的小瓶中,磁力搅拌,直至所有组分完全溶解。
[0119]
实验结果表明,处方8、9和11无法形成均一透明的油溶液,处方6、7和10形成了均一透明的油溶液,并且观察到这些油溶液放置后均不发生分层。
[0120]
将按照处方6-11制备的浓缩液以1g:100ml的比例用5%葡萄糖注射液稀释后,均形成白色乳剂,将乳剂在室温下放置24小时,用激光粒度仪(美国pss,型号nicompz3000)测定其平均粒径,结果如表5所示。
[0121]
表5—用复合乳化剂制备的浓缩液稀释后形成的乳剂的粒径和稳定性
[0122][0123]
“‑‑”
表示因产生分层现象未测量粒径;“md”表示平均粒径(mean diameter),单位是nm;“pdi”表示粒径分散系数(polydispersity index)。
[0124]
如表5所示,将上述乳剂在室温下放置24小时,由包含hs15与epc组成的复合乳化剂的处方6以及包含hs15与spc组成的复合乳化剂的处方7制备的油溶液所形成的乳剂粒径无明显变化;其余乳剂粒径较大且均不稳定,在形成乳剂后2h左右发生分层,无法满足静脉注射给药的需求。
[0125]
由此说明,应当使用包含磷脂和非磷脂复合乳化剂的处方,且其中的磷脂不应当是hspc。
[0126]
实施例2:油的筛选
[0127]
发明人对多种可药用的油进行了考察,具体实验设计如下:
[0128]
表6—包含不同油的处方
[0129][0130]
工艺:将处方量的各组分加入20ml的小瓶中,磁力搅拌,直至所有组分完全溶解。
[0131]
观察按照处方12-21制备的浓缩液的外观。将按照处方12-21制备的浓缩液以1g:100ml的比例用5%葡萄糖注射液稀释,并将所得乳剂在室温下放置24小时,观察浓缩液形成乳剂的情况、测定所得乳剂的粒径并考察其稳定性。结果如表7所示。
[0132]
表7—用处方12-21制备的浓缩液及其稀释产生的乳剂的稳定性
[0133][0134]“—”:由于浓缩液是浑浊的液体,在室温放置约1h即分层,不是稳定的体系,不再考察
[0135]
由表7的实验结果可见,只有按照包含中链甘油三酸酯的处方12所制备的浓缩液能形成均一透明的油溶液,并且在稀释后形成稳定的乳剂。按照包含大豆油、橄榄油、鱼油、玉米油、结构油、蓖麻油、葵花籽油、棉籽油、茶油等其它注射用油的处方13-21所制备的浓缩液均为浑浊的液体,无法形成均一透明的油溶液,在室温放置1h左右便会出现分层现象。因此,应使用mct作为油。
[0136]
实施例3:塞来昔布浓缩液中的助乳化剂的筛选
[0137]
发明人对多种可药用的助乳化剂进行了考察,具体实验设计如下:
[0138]
表8—包含不同助乳化剂的处方
[0139]
组分处方22处方23处方24处方25处方26处方27塞来昔布0.5g0.5g0.5g0.5g0.5g0.5ghs153.0g3.0g3.0g3.0g3.0g3.0gepc0.3g0.3g0.3g0.3g0.3g0.3gmct4.0g4.0g4.0g4.0g4.0g4.0g无水乙醇1.6g
ꢀꢀꢀꢀꢀ
丙二醇 1.6g
ꢀꢀꢀꢀ
甘油
ꢀꢀ
1.6g
ꢀꢀꢀ
peg200
ꢀꢀꢀ
1.6g
ꢀꢀ
peg300
ꢀꢀꢀꢀ
1.6g peg400
ꢀꢀꢀꢀꢀ
1.6g总计10g10g10g10g10g10g
[0140]
工艺:将处方量的各组分加入20ml的小瓶中,磁力搅拌,直至所有组分完全溶解。
[0141]
观察按照处方22-27制备的浓缩液的外观。将按照处方22-27制备的浓缩液以1g:100ml的比例用5%葡萄糖注射液稀释,并将所得乳剂在室温下放置24小时,观察浓缩液形成乳剂的情况、测定所得乳剂的粒径并考察其稳定性。结果如表9所示。
[0142]
表9—用处方22-27制备的浓缩液及其稀释产生的乳剂的稳定性
[0143][0144]“—”:由于浓缩液是浑浊的液体,在室温放置约1h即分层,不是稳定的体系,不再考察
[0145]
由表9的实验结果可见,只有按照包含无水乙醇的处方22所制备的浓缩液能形成均一透明的油溶液,并且在稀释后能形成稳定的乳剂。按照包含丙二醇、甘油、peg200、peg300、peg400等其它助乳化剂的处方23-27制备的浓缩液均为浑浊的液体,在室温放置1h左右便会出现分层现象。因此,应使用无水乙醇作为助乳化剂。
[0146]
实施例4:含有难溶性药物的浓缩液的制备以及由其形成的乳剂的粒径和稳定性研究(*将难溶性药物、复合乳化剂、油、助乳化剂的总量视为100%)
[0147]
浓缩液1
[0148]
处方:
[0149]
组分重量百分比*用量塞来昔布5%0.5gmct28%2.8ghs1548%4.8gepc2.8%0.28g无水乙醇16.2%1.62g
共计 10g
[0150]
工艺:
[0151]
称取处方量的epc与塞来昔布,放入20ml小瓶中,加入处方量的无水乙醇,在60℃的水浴中以2000rpm搅拌1min。再向其中加入处方量的mct、hs15,在60℃的水浴中以2000rpm搅拌5min,获得均一透明的油溶液。
[0152]
乳剂稳定性考察:
[0153]
将浓缩液1以1g:100ml的比例用5%葡萄糖注射液稀释,测定所得乳剂的粒径并考察其24小时的稳定性。
[0154][0155]
浓缩液2
[0156]
处方:
[0157]
组分重量百分比*用量塞来昔布5%0.5gmct28%2.8ghs1548%4.8gepc3%0.3g无水乙醇16%1.59g枸橼酸 0.01g共计 10g
[0158]
工艺:
[0159]
称取处方量的epc,放入到20ml小瓶中,加入处方量的无水乙醇,在60℃的水浴中以2000rpm搅拌1min溶解。再向其中加入处方量的塞来昔布,在60℃的水浴中以2000rpm搅拌1min。再向其中加入处方量的hs15、mct和枸橼酸,在60℃的水浴中以2000rpm搅拌5min,获得均一透明的油溶液。
[0160]
乳剂稳定性考察:
[0161]
将浓缩液2以1g:100ml的比例用0.9%氯化钠注射液稀释,测定所得乳剂的粒径并考察其24小时的稳定性。
[0162][0163]
浓缩液3
[0164]
处方:
[0165][0166][0167]
工艺:
[0168]
称取处方量的epc,放入20ml小瓶中,加入处方量的无水乙醇,在60℃的水浴中以2000rpm搅拌1min溶解。再向其中加入处方量的紫杉醇,在60℃的水浴中以2000rpm搅拌1min。再向其中加入处方量的hs15和mct,在60℃的水浴中以2000rpm搅拌5min,获得均一透明的油溶液。
[0169]
乳剂稳定性考察:
[0170]
将浓缩液3以1g:50ml的比例用5%葡萄糖注射液稀释,测定所得乳剂的粒径并考察其24小时的稳定性。
[0171]
指标0h24h外观白色乳剂白色乳剂,未见药物析出平均粒径(nm)255.34257.46pdi0.210.25
[0172]
浓缩液4
[0173]
处方:
[0174]
组分重量百分比*用量多西他赛2%0.2gmct28%2.8ghs1548%4.8gepc3%0.3g
无水乙醇19%1.9g共计 10g
[0175]
工艺:
[0176]
称取处方量的epc、多西他赛、无水乙醇、mct、hs15,放入20ml小瓶中,在60℃的水浴中以2000rpm搅拌5min,获得均一透明的油溶液。
[0177]
乳剂稳定性考察:
[0178]
将浓缩液4以1g:50ml的比例用5%葡萄糖注射液稀释,测定所得乳剂的粒径并考察其24小时的稳定性。
[0179][0180]
浓缩液5
[0181]
处方:
[0182]
组分重量百分比*用量布洛芬10%1.0gmct23%2.3ghs1548%4.8gepc3%0.3g无水乙醇16%1.6g共计 10g
[0183]
工艺:
[0184]
称取处方量的epc、布洛芬、无水乙醇、mct、hs15,放入20ml小瓶中,在60℃的水浴中以2000rpm搅拌5min,获得均一透明的油溶液。
[0185]
乳剂稳定性考察:
[0186]
将浓缩液5以1g:100ml的比例用5%葡萄糖注射液稀释,测定所得乳剂的粒径并考察其24小时的稳定性。
[0187][0188]
浓缩液6
[0189]
处方:
[0190]
组分重量百分比*用量尼莫地平0.1%0.01gmct37%3.7ghs1543.9%4.39gepc3%0.3g无水乙醇16%1.6g共计 10g
[0191]
工艺:
[0192]
称取处方量的epc、尼莫地平、无水乙醇、mct、hs15,放入20ml小瓶中,在60℃的水浴中以2000rpm搅拌5min,获得均一透明的油溶液。
[0193]
乳剂稳定性考察:
[0194]
将浓缩液6以1g:50ml的比例用5%葡萄糖注射液稀释,测定所得乳剂的粒径并考察其24小时的稳定性。
[0195][0196]
浓缩液7
[0197]
处方:
[0198]
组分重量百分比*用量辅酶q105%0.5gmct28%2.8gtpgs48%4.8gepc1%0.1g无水乙醇18%1.8g共计 10g
[0199]
工艺:
[0200]
称取处方量的epc、辅酶q10、无水乙醇、mct、tpgs,放入20ml小瓶中,在60℃的水浴中以2000rpm搅拌5min,获得均一透明的油溶液。
[0201]
乳剂稳定性考察:
[0202]
将浓缩液7以1g:100ml的比例用5%葡萄糖注射液稀释,测定所得乳剂的粒径并考察其24小时的稳定性。
[0203][0204]
浓缩液8
[0205]
处方:
[0206]
组分重量百分比*用量卡巴他赛2%0.2gmct32%3.2ghs1547%4.7gepc5%0.5g无水乙醇14%1.4g共计 10g
[0207]
工艺:
[0208]
按顺序称取处方量的epc、卡巴他赛、无水乙醇、mct、hs15,放入20ml小瓶中,在60℃的水浴中以2000rpm搅拌5min,获得均一透明的油溶液。
[0209]
乳剂稳定性考察:
[0210]
将浓缩液8以1g:50ml的比例用5%葡萄糖注射液稀释,测定所得乳剂的粒径并考察其24小时的稳定性。
[0211][0212]
浓缩液9
[0213]
处方:
[0214]
组分重量百分比*用量依托考昔5%0.5gmct30%3.0gtween 8046%4.6gspc3%0.3g无水乙醇16%1.6g共计 10g
[0215]
工艺:
[0216]
称取处方量的spc与依托考昔,放入20ml小瓶中,加入处方量的无水乙醇,在60℃的水浴中以2000rpm搅拌1min。再向其中加入处方量的mct和tween80,在60℃的水浴中以2000rpm搅拌5min,获得均一透明的油溶液。
[0217]
乳剂稳定性考察:
[0218]
将浓缩液9以1g:100ml的比例用5%葡萄糖注射液稀释,测定所得乳剂的粒径并考察其24小时的稳定性。
[0219][0220]
浓缩液10
[0221]
处方:
[0222]
组分重量百分比*用量泊沙康唑1%0.1gmct25%2.5grh4054%5.4gspc3%0.3g无水乙醇17%1.7g共计 10g
[0223]
工艺:
[0224]
称取处方量的spc,放入20ml小瓶中,加入处方量的无水乙醇,在60℃的水浴中以2000rpm搅拌1min溶解。再向其中加入处方量的泊沙康唑,在60℃的水浴中以2000rpm搅拌1min。再向其中加入处方量的rh40和mct,在60℃的水浴中以2000rpm搅拌5min,获得均一透明的油溶液。
[0225]
乳剂稳定性考察:
[0226]
将浓缩液10以1g:50ml的比例用5%葡萄糖注射液稀释,测定所得乳剂的粒径并考察其12小时的稳定性。
[0227][0228]
浓缩液11
[0229]
处方:
[0230]
组分重量百分比*用量环孢素2%0.2gmct33%3.3gelp40%4.0gepc5%0.5g无水乙醇20%2.0g共计 10g
[0231]
工艺:
[0232]
称取处方量的epc,放入20ml小瓶中,加入处方量的无水乙醇,在60℃的水浴中以2000rpm搅拌1min直至溶解。再向其中加入处方量的环孢素,在60℃的水浴中以2000rpm搅拌1min。再向其中加入处方量的elp和mct,在60℃的水浴中以2000rpm搅拌5min,获得均一透明的油溶液。
[0233]
乳剂稳定性考察:
[0234]
将浓缩液11以1g:50ml的比例用5%葡萄糖注射液稀释,测定所得乳剂的粒径并考察其24小时的稳定性。
[0235][0236]
浓缩液12
[0237]
处方:
[0238]
组分重量百分比*用量氟比洛芬酯1%0.1gmct28%2.8ghs1549%4.9gepc3%0.3g无水乙醇19%1.9g共计 10g
[0239]
工艺:
[0240]
按顺序称取处方量的epc、氟比洛芬酯、无水乙醇、mct、hs15,放入20ml小瓶中,在60℃的水浴中以2000rpm搅拌5min,获得均一透明的油溶液。
[0241]
乳剂稳定性考察:
[0242]
将浓缩液12以1g:50ml的比例用5%葡萄糖注射液稀释,测定所述乳剂的粒径并考察其24小时的稳定性。
[0243][0244]
浓缩液13
[0245]
处方:
[0246]
组分重量百分比*用量右旋布洛芬10%1.0gmct23%2.3gelp48%4.8gspc3%0.3g无水乙醇16%1.6g共计 10g
[0247]
工艺:
[0248]
按顺序称取处方量的spc、右旋布洛芬、无水乙醇、mct、elp,放入20ml小瓶中,在60℃的水浴中以2000rpm搅拌5min,获得均一透明的油溶液。
[0249]
乳剂稳定性考察:
[0250]
将浓缩液13以1g:100ml的比例用5%葡萄糖注射液稀释,测定所得乳剂的粒径并考察其24小时的稳定性。
[0251][0252]
浓缩液14
[0253]
处方:
[0254]
组分重量百分比*用量左西孟旦0.1%0.01gmct37%3.7ghs1543.9%4.39gepc3%0.3g无水乙醇16%1.6g共计 10g
[0255]
工艺:
[0256]
按顺序称取处方量的epc、左西孟旦、无水乙醇、mct、hs15,放入20ml小瓶中,在60℃的水浴中以2000rpm搅拌5min,获得均一透明的油溶液。
[0257]
乳剂稳定性考察:
[0258]
将浓缩液14以1g:50ml的比例用5%葡萄糖注射液稀释,测定所得乳剂的粒径并考察其24小时的稳定性。
[0259][0260]
浓缩液15
[0261]
处方:
[0262]
组分重量百分比*用量丁酸氯维地平2%0.2gmct28%2.8ghs1548%4.8gepc4%0.4g无水乙醇17.9%1.79g棕榈酸抗坏血酸酯 0.01g共计 10g
[0263]
工艺:
[0264]
按顺序称取处方量的epc、丁酸氯维地平、无水乙醇、mct、hs15,放入20ml小瓶中,在60℃的水浴中以2000rpm搅拌5min,获得均一透明的油溶液。
[0265]
乳剂稳定性考察:
[0266]
将浓缩液15以1g:50ml的比例用5%葡萄糖注射液稀释,测定所得乳剂的粒径并考察其24小时的稳定性。
[0267][0268]
浓缩液16
[0269]
处方:
[0270]
组分重量百分比*用量氯吡格雷2%0.2g
mct32%3.2ghs1547%4.7gepc5%0.5g无水乙醇14%1.4g共计 10g
[0271]
工艺:
[0272]
按顺序称取处方量的epc、氯吡格雷、无水乙醇、mct、hs15,放入20ml小瓶中,在60℃的水浴中以2000rpm搅拌5min,获得均一透明的油溶液。
[0273]
乳剂稳定性考察:
[0274]
将浓缩液16以1g:100ml的比例用5%葡萄糖注射液稀释,测定所得乳剂的粒径并考察其24小时的稳定性。
[0275][0276]
浓缩液17
[0277]
处方:
[0278]
组分重量百分比*用量瑞德西韦1%0.1gmct37%3.7ghs1543%4.3gepc3%0.3g无水乙醇16%1.6g共计 10g
[0279]
工艺:
[0280]
称取处方量的epc与瑞德西韦,放入20ml小瓶中,加入处方量的无水乙醇,在60℃的水浴中以2000rpm搅拌1min。再向其中加入处方量的mct和hs15,在60℃的水浴中以2000rpm搅拌5min,获得均一透明的油溶液。
[0281]
乳剂稳定性考察:
[0282]
将浓缩液17以1g:100ml的比例用5%葡萄糖注射液稀释,测定所得乳剂的粒径并考察其24小时的稳定性。
[0283][0284]
浓缩液18
[0285]
处方:
[0286]
组分重量百分比*用量他克莫司1%0.1gmct28%2.8ghs1550%5.0gepc3%0.3g无水乙醇18%1.8g共计 10g
[0287]
工艺:
[0288]
称取处方量的epc,放入20ml小瓶中,加入处方量的无水乙醇,在60℃的水浴中以2000rpm搅拌1min溶解。再向其中加入处方量的他克莫司,在60℃的水浴中以2000rpm搅拌1min。再向其中加入处方量的hs15和mct,在60℃的水浴中以2000rpm搅拌5min,获得均一透明的油溶液。
[0289]
乳剂稳定性考察:
[0290]
将浓缩液18以1g:50ml的比例用5%葡萄糖注射液稀释,测定所得乳剂的粒径并考察其24小时的稳定性。
[0291][0292][0293]
浓缩液19
[0294]
处方:
[0295]
组分重量百分比*用量紫杉醇2%0.2gmct40%4.0gelp33%3.3gepc5%0.5g
无水乙醇20%2.0g共计 10g
[0296]
工艺:
[0297]
称取处方量的epc,放入20ml小瓶中,加入处方量的无水乙醇,在60℃的水浴中以2000rpm搅拌1min溶解。再向其中加入处方量的紫杉醇,在60℃的水浴中以2000rpm搅拌1min。再向其中加入处方量的elp和mct,在60℃的水浴中以2000rpm搅拌5min,获得均一透明的油溶液。
[0298]
乳剂稳定性考察:
[0299]
将浓缩液19以1g:50ml的比例用5%葡萄糖注射液稀释,测定所得乳剂的粒径并考察其24小时的稳定性。
[0300][0301]
浓缩液20
[0302]
处方:
[0303][0304][0305]
工艺:
[0306]
按顺序称取处方量的epc、布洛芬、无水乙醇、mct、hs15,放入20ml小瓶中,在60℃的水浴中以2000rpm搅拌5min,获得均一透明的油溶液。
[0307]
乳剂稳定性考察:
[0308]
将浓缩液20以1g:50ml的比例用5%葡萄糖注射液稀释,测定所得乳剂的粒径并考察其24小时的稳定性。
[0309][0310]
浓缩液21
[0311]
处方:
[0312]
组分重量百分比*用量卡铂1%0.1gmct32%3.2gtween 8048%4.8gspc3%0.3g无水乙醇16%1.6g共计 10g
[0313]
工艺:
[0314]
按顺序称取处方量的spc、卡铂、无水乙醇、mct、tween 80,放入到20ml小瓶中,在60℃的水浴中以2000rpm搅拌5min,获得均一透明的油溶液。
[0315]
乳剂稳定性考察:
[0316]
将浓缩液21以1g:50ml的比例用5%葡萄糖注射液稀释,测定所得乳剂的粒径并考察其24小时的稳定性。
[0317][0318]
浓缩液22
[0319]
处方:
[0320]
组分重量百分比*用量地塞米松棕榈酸酯1%0.1gmct37%3.7ghs1543%4.3gepc3%0.3g无水乙醇16%1.6g共计 10g
[0321]
工艺:
[0322]
按顺序称取处方量的epc、地塞米松棕榈酸酯、无水乙醇、mct、hs15,加入20ml小瓶中,在60℃的水浴中以2000rpm搅拌5min,获得均一透明的油溶液。
[0323]
乳剂稳定性考察:
[0324]
将浓缩液22以1g:50ml的比例用5%葡萄糖注射液稀释,测定所得乳剂的粒径并考察其24小时的稳定性。
[0325][0326]
浓缩液23
[0327]
处方:
[0328]
组分重量百分比*用量伐地昔布5%0.5gmct28%2.8ghs1548%4.8gepc1%0.1g无水乙醇18%1.8g共计 10g
[0329]
工艺:
[0330]
按顺序称取处方量的epc、伐地昔布、无水乙醇、mct、hs15,放入20ml小瓶中,在60℃的水浴中以2000rpm搅拌5min,获得均一透明的油溶液。
[0331]
乳剂稳定性考察:
[0332]
将浓缩液23以1g:50ml的比例用5%葡萄糖注射液稀释,测定所得乳剂的粒径并考察其24小时的稳定性。
[0333][0334]
浓缩液24
[0335]
处方:
[0336]
组分重量百分比*用量伏立康唑6%0.6gmct30%3.0g
hs1546%4.6gepc4%0.4g无水乙醇14%1.4g共计 10g
[0337]
工艺:
[0338]
按顺序称取处方量的epc、伏立康唑、无水乙醇、mct、hs15,放入20ml小瓶中,在60℃的水浴中以2000rpm搅拌5min,获得均一透明的油溶液。
[0339]
乳剂稳定性考察:
[0340]
将浓缩液24以1g:50ml的比例用5%葡萄糖注射液稀释,测定所得乳剂的粒径并考察其24小时的稳定性。
[0341][0342]
浓缩液25
[0343]
处方:
[0344]
组分重量百分比*用量阿奇霉素12%1.2gmct24%2.4ghs1548%4.8gepc2%0.2g无水乙醇14%1.4g共计 10g
[0345]
工艺:
[0346]
按顺序称取处方量的epc、阿奇霉素、无水乙醇、mct、hs15,放入20ml小瓶中,在60℃的水浴中以2000rpm搅拌5min,获得均一透明的油溶液。
[0347]
乳剂稳定性考察:
[0348]
将浓缩液25以1g:50ml的比例用5%葡萄糖注射液稀释,测定所得乳剂的粒径并考察其24小时的稳定性。
[0349]
指标0h24h外观白色乳剂白色乳剂,未见药物析出平均粒径(nm)125.56130.13pdi0.140.18
[0350]
浓缩液26
[0351]
处方:
[0352]
组分重量百分比*用量丙泊酚6%0.6gmct35%3.5ghs1548%4.8gepc2%0.2g无水乙醇9%0.9g共计 10g
[0353]
工艺:
[0354]
将处方量的各组分加入20ml小瓶中,磁力搅拌,直至所有组分完全溶解,得到均一透明的油溶液。
[0355]
乳剂稳定性考察:
[0356]
将浓缩液26以1g:6ml的比例用5%葡萄糖注射液稀释,测定所得乳剂的粒径并考察其24小时的稳定性。
[0357]
指标0h24h外观白色乳剂白色乳剂,未见药物析出平均粒径(nm)212.16220.38pdi0.110.15
[0358]
浓缩液27
[0359]
处方:
[0360]
组分重量百分比*用量紫杉醇1.5%0.15gmct29.4%2.93ghs1549.1%4.9gepc3%0.3g无水乙醇17%1.7g枸橼酸 0.02g共计 10g
[0361]
工艺:
[0362]
称取处方量的epc,放入20ml小瓶中,加入处方量的无水乙醇,在60℃的水浴中以2000rpm搅拌1min溶解。再向其中加入处方量的紫杉醇,在60℃的水浴中以2000rpm搅拌1min。再向其中加入处方量的hs15和mct,在60℃的水浴中以2000rpm搅拌5min,获得均一透明的油溶液。
[0363]
乳剂稳定性考察:
[0364]
将浓缩液27以1g:100ml的比例用5%葡萄糖注射液稀释,测定所得乳剂的粒径并考察其24小时的稳定性。
[0365]
指标0h24h外观白色乳剂白色乳剂,未见药物析出平均粒径(nm)93.2093.85
pdi0.260.27
[0366]
实施例5:本发明的浓缩液的稳定性考察
[0367]
按照《中国药典》2015版四部通则9001中关于原料药与药物制剂稳定性试验指导原则,对实施例4中的浓缩液1进行加速稳定性考察。
[0368]
1、供试品:实施例4中制备的浓缩液1,批号2020041501
[0369]
2、稳定性试验放样条件:
[0370][0371]
3、分析方法
[0372]
(1)性状
[0373]
检查方法:目测。
[0374]
(2)含量
[0375]
检测方法:hplc法
[0376]
色谱条件:色谱柱:苯基键合硅胶为填充剂,250mm
×
4.6mm,5um;流动相:2.7g/l磷酸二氢钾溶液(用磷酸调ph至3.0)-甲醇-乙腈(60:30:10);检测波长:215nm;柱温:60℃;流速:1.5ml/min;进样量:20ul;运行时间:30min。
[0377]
具体试验操作:精密称取塞来昔布杂质a(4-[5-(3-甲基苯基)-3-9三氟甲基)-1h-吡唑-1-基]苯磺酰胺)、杂质b(4-[3-(4-甲基苯基)-5-(三氟甲基)-1h-吡唑-1-基)苯磺酰胺)各12mg于同一个100ml容量瓶中,用甲醇溶解并稀释至刻度,摇匀,作为系统对照溶液。精密称取塞来昔布标准品25mg于50ml容量瓶中,加入1ml系统对照溶液,用甲醇溶解并稀释至刻度,摇匀,作为系统适用性溶液。精密称取塞来昔布标准品25mg,用甲醇溶解并稀释至刻度,摇匀,作为对照品溶液。精密称取供试品(实施例4中的浓缩液1)1.0g于100ml容量瓶中,用甲醇溶解并稀释至刻度,摇匀,作为供试品溶液。分别量取用0.45μm尼龙膜过滤的对照品溶液与供试品溶液各20μl,注入液相色谱仪,记录色谱图,按外标法以峰面积计算塞来昔布和杂质的含量。
[0378]
(3)乳剂的性状、粒径、ph值和稳定性
[0379]
取1g浓缩液1,加入到100ml 5%葡萄糖注射液中,手动摇匀后,得到乳剂;用激光粒度仪(pss nicomp z3000)测定所得乳剂的平均粒径和多分散系数(pdi);用ph计测定乳剂的ph值;室温静置24h,观察是否有药物析出。
[0380]
4、实验结果
[0381]
表10—实施例4的浓缩液1的稳定性
[0382][0383]
表10中的实验结果显示,浓缩液1在40℃
±
2℃、rh60%
±
5%下放置6个月具有良好的物理和化学稳定性。
[0384]
表11—由实施例4的浓缩液1制备的乳剂的稳定性
[0385][0386][0387]
表11中的实验结果显示,在6个月的稳定性试验期间,浓缩液1经5%葡萄糖注射液稀释后均能形成乳剂,并且所得乳剂在24h内平均粒径、pdi、ph值均保持稳定,未发生分层或药物析出,具有良好的稳定性。
[0388]
实施例6:含有丙泊酚的浓缩液的制备及其性质
[0389]
在本实施例中,比较了本技术的实施例4中的浓缩液26与中国专利申请cn10834851a的丙泊酚-浓缩物b6、市售的丙泊酚中/长链脂肪乳(批号:16mc0288,北京费森北京费森尤斯卡比医药有限公司)。
[0390]
本技术的实施例4中的浓缩液26与中国专利申请cn10834851a的丙泊酚-浓缩物b6的组成如下:
[0391]
表12-cn10834851a的丙泊酚-浓缩物b6与本技术的浓缩液26的处方
[0392]
组分cn10834851a的丙泊酚-浓缩物b6本技术的浓缩液26丙泊酚0.6g0.6ghs157.0246g4.8gepc0.188g0.2gmct0.752g3.5g乙醇0.4954g0.9g丙二醇0.188g
‑‑
peg4000.752g
‑‑
总计10g10g
[0393]
工艺:将每种处方中规定量的各组分加入20ml的小瓶中,磁力搅拌,直至所有组分完全溶解。
[0394]
所得的两种丙泊酚浓缩液均为均一透明的油溶液。
[0395]
将所得油溶液以1g:6ml的比例用5%葡萄糖注射液稀释,至丙泊酚浓度为10mg/ml。
[0396]
用激光粒度仪(美国pss,型号nicompz3000)测定其平均粒径和pdi;采用3kd超滤管(密理博)离心法分离游离药物,hplc法(仪器型号:lc-20at岛津;色谱柱:agilent zorbaxextend c18(250mm
×
4.6mm,5um);流动相:磷酸二氢钠溶液(ph3.0)-乙腈);检测波长:275nm;柱温:40℃;流速:1.0ml/min;进样量:10ul;测定游离药物浓度。
[0397]
此外,采用体外溶血法(2%家兔红细胞混悬液)测定溶血率。具体实验方案如下:
[0398]
阴性对照管的配制
[0399]
取试管,加入2%红细胞混悬液2.0ml和5%葡萄糖注射液2.0ml。
[0400]
阳性对照管的配制
[0401]
取试管,加入2%红细胞混悬液2.5ml,离心(1500r/min)5min,弃去上层无色溶液1.5ml,剩余物加蒸馏水4ml使红细胞破裂,得完全溶血的阳性对照管。
[0402]
供试品管的配制
[0403]
取试管,加入0.3ml丙泊酚浓缩液,加入5%葡萄糖注射液2.2ml和2%红细胞混悬液2.5ml,摇匀,作为供试品管。供试品一式两份地配制。
[0404]
样品测定
[0405]
将各试管置于37
±
0.5℃的水浴中温育,3h后取出,离心(1500r/min)5min,精密量取上清液0.2ml,精密加入破乳剂(移取浓盐酸0.2ml,加入无水乙醇79.8ml,混匀,即得)3.2ml(破乳比16:1),摇匀,取适量,在400nm处测定血红素吸光度(仪器:紫外分光光度计,型号:t6新世纪,厂家:北京普西通用仪器有限责任公司;波长:400nm),按照以下公式计算溶血百分比:
[0406]
溶血百分比(%)=(供试品管吸光度-阴性对照管吸光度)/(阳性对照管吸光度-阴性对照管吸光度)
×
100%
[0407]
实验结果如表13所示。
[0408]
表13-cn10834851a的丙泊酚-浓缩物b6、本技术的浓缩液26和市售丙泊酚脂肪乳的性质比较
[0409][0410]
由表13中的实验结果可见,cn10834851a的丙泊酚-浓缩物b6经稀释后所得到的乳剂是均一透明的溶液,乳剂的粒径只有16.88nm,类似于真溶液,并且在室温放置24h保持稳定。乳剂中游离丙泊酚的量高达0.6%,导致其体外溶血率高达31.62%。
[0411]
本技术的浓缩液28经稀释后形成白色均一的乳剂,其平均粒径为212.16nm,并且在室温放置24h保持稳定。乳剂中游离丙泊酚的量仅有0.01%,体外溶血率仅为2.25%。
[0412]
在本领域中已知的是,制剂中游离丙泊酚的量是引起注射疼痛的主要原因,也是引起体外溶血的主要原因,由丙泊酚引起的溶血率在3.1%~58%范围内与游离药物浓度具有良好的线性关系(r=0.9650)(例如,参见,蔡伟惠和金方,“不同丙泊酚载药体系中游离药物浓度与体外溶血性的关系”,中国医药工业杂志,2008,39(1)22~26)。因此,与cn10834851a的丙泊酚-浓缩物b6相比,本技术的浓缩液26减少了辅料的种类,同时保持了良好的稳定性,而且具有更少的副作用(即,注射疼痛更小、溶血率更低)。
[0413]
此外,本技术的浓缩液26经稀释后形成的乳剂在外观、平均粒径、pdi及溶血率方面均与市售丙泊酚中/长链脂肪乳注射液类似,完全符合静脉内注射给药的要求,同时避免了脂肪乳注射剂的复杂制备工艺和严格的储存和运输条件。
[0414]
实施例7:塞来昔布浓缩液大鼠足底切口痛药效学考察
[0415]
1.实验材料
[0416]
1.1供试品
[0417]
名称:塞来昔布注射液
[0418]
配制方法:用2.5ml的注射器抽取2.1g实施例4中制备的浓缩液2(批号2020060401),将其注入100ml 5%葡萄糖注射液中,手动摇匀,即得乳剂。所得乳剂的性质如下:
[0419]
塞来昔布浓度mdpdi1.05mg/ml33.42nm0.03
[0420]
1.2阳性对照
[0421]
(1)阳性对照一
[0422]
名称:帕瑞昔布钠注射液
[0423]
批号:cj2725
[0424]
规格:40mg/支
[0425]
性状:白色冻干粉饼
[0426]
有效期:2022年03月
[0427]
生产厂家:辉瑞
[0428]
阳性对照一的配制:用5%葡萄糖注射液稀释至帕瑞昔布钠的浓度为0.41mg/ml。
[0429]
(2)阳性对照二
[0430]
名称:氟比洛芬酯注射液(凯纷)
[0431]
批号:3e309e
[0432]
规格:5ml:50mg
[0433]
剂型:注射液
[0434]
有效期:2021年03月20日
[0435]
生产厂家:北京泰德制药
[0436]
阳性对照二的配制:用5%葡萄糖注射液稀释至氟比洛芬酯的浓度为1.05mg/ml。
[0437]
1.3阴性对照
[0438]
名称:5%葡萄糖注射液
[0439]
批号:1909061b
[0440]
厂家:安徽双鹤药业有限责任公司。
[0441]
1.4空白制剂
[0442]
名称:空白制剂(与供试品的差别仅在于,将难溶性药物替换为相同重量的无水乙醇,其它组分和配制方法与供试品完全一致)
[0443]
批号:2020060901
[0444]
规格:0mg/瓶
[0445]
浓度:0mg/ml
[0446]
性状:澄清透明溶液
[0447]
生产厂家:北京德立福瑞医药科技有限公司
[0448]
1.5实验动物
[0449]
品种:sd大鼠
[0450]
级别:spf级动物
[0451]
性别和数量:雄性,60只
[0452]
动物年龄:约6~8周龄
[0453]
动物体重:220~260g
[0454]
动物来源:北京华阜康生物科技股份有限公司
[0455]
2.实验设计
[0456]
将市售帕瑞昔布钠注射液和氟比洛芬酯注射液(凯纷)用于人的给药量折算至炎症模型动物(大鼠)的给药量,确定其对大鼠的给药剂量分别是8.4mg/kg和21mg/kg。
[0457]
将供试品、两种阳性对照、阴性对照和空白制剂按照表14中所给出的浓度和剂量静脉注射给药1次。
[0458]
2.1大鼠足底切口模型的建立
[0459]
足底切口模型:取1.4中所述的雄性sd大鼠,在实验室条件下饲养1周,然后,使大鼠吸入异氟烷麻醉后,从足底近端0.5cm处向趾部作一长约1cm的切口:切开皮肤、筋膜后,用眼科镊挑起足底肌肉并纵向切割(保持肌肉的起止及附着完善);按压止血。在各动物的足底切口附近进行皮下注射生理盐水及各相应的受试药物。之后以热板方法进行痛阈检测。该大鼠模型用于模拟临床术后疼痛。
[0460]
2.2剂量和分组
[0461]
将造模成功的sd大鼠随机分为5组,每组10只。分别为帕瑞昔布钠组(a组)、塞来昔布注射液组(b组)、氟比洛芬酯注射液(凯纷)组(c组)、阴性对照组(d组)和空白制剂组(e组)。各组分别给予对应的制剂,给药方式为尾静脉滴注,给药1次。给药方案细节参见表14。
[0462]
表14—sd大鼠药效试验剂量设计
[0463][0464]
注:(1)d组为阴性对照组,给予大鼠5%葡萄糖注射液;(2)e组为空白制剂组,给予大鼠塞来昔布注射液空白制剂稀释后的乳剂(即,与b组相同的乳剂,但是不含有塞来昔布,塞来昔布的重量份数以无水乙醇代替);(3)各组动物的给药方式均为静脉滴注,采用注射泵静脉滴注,滴注时间为25min~30min。
[0465]
2.3检测指标频率及方法
[0466]
感觉阻滞的评价:对于感觉阻滞的评价,采用的是弗莱毛(von frey hair,stoelting,wood dale,美国),具体操作同动物筛选和分组项下测定痛阈值的方法,弗莱毛测试给药后30min、1h、2h、4h、8h测定给药侧。
[0467]
在环境温度(22
±
1℃)的安静环境中,采用弗莱毛细丝测定机械性痛觉超敏来评定大鼠疼痛状况。
[0468]
测定时将大鼠置于底为网格的特制有机玻璃格子(26cm
×
20cm
×
14cm)或钢丝网内,限制其在一个小范围内自由活动,适应20min后,根据dixon介绍的up and down法,将一系列von frey细丝(0.4、0.6、1.4、2.0、4.0、6.0、8.0、15.0g)从2.0g力度开始刺激大鼠移植侧脚掌中部皮肤,接触足底加压使丝呈c状,弧度如3/8缝合针,计时,至动物抬脚或走开。观察大鼠缩足反应。
[0469]
无反应记为阴性反应“o”,有反应(缩足或舔足)记为阳性反应“x”。如果第一根毛刺激没有反应,则给予大一级力度的毛刺激;如果有反应则改用小一级力度的毛刺激,反复类推,如此连续进行,直至出现第1次阳性和阴性(或阴性和阳性)反应的骑跨,再向下连续测定4次。以出现“x”的前一次的“o”作为起点,选择包括该起点范围内的连续6次刺激所得到的ox值作为推算50%缩足反应阈值的关键序列。但也不完全限于6次(最少4次,最多9次),不同刺激之间相隔30s,以消除前一刺激的影响,若直至测试完最大力度(15.10g)的von frey纤维,动物仍不出现缩足反应,则大鼠的50%缩足阈值为15.00g。得到一串以“o”或“x”组合的序列后,将该序列和最后一根毛的力度(f)输入公式计算出机械性疼痛阈值作为机械性痛阈值。
[0470]
50%缩足反应阈值(g)=(10[xf+kδ])/10 000
[0471]
其中,
[0472]
xf=log(f*10000);
[0473]
δ为各个毛力度取log后的均差,在此约等于0.224;
[0474]
k为根据测量所得“x”、“o”序列查表后得到的值。
[0475]
为避免频繁或长时间的刺激造成动物耐受或痛敏,每根毛每次刺激时间最长不超过8s,实验中间隔4d以上测量一次机械性痛觉超敏。
[0476]
3.实验结果
[0477]
表15—各组的50%缩足反应阈值
[0478][0479]
将表15中的数据绘图,得到图1。在图1中,第一条曲线(
▲
)代表塞来昔布乳剂的实验结果,第二条曲线(
■
)是代表氟比洛芬酯注射液的实验结果,第三条曲线(
●
)代表帕瑞昔布钠注射液的实验结果,第四条曲线(
○
)代表空白制剂的实验结果,第五条曲线代表阴性对照(5%葡萄糖注射液)的实验结果。由图1可以清楚地看到,在大鼠足底切口痛模型中,与市售的帕瑞昔布钠注射液及氟比洛芬酯注射液,本发明的塞来昔布乳剂注射液镇痛效果更强,具有显著性差异(采用graphpad prism软件进行one-way analysis of variance(anova)检验,***表示本发明的塞来昔布乳剂注射液与帕瑞昔布钠注射液相比具有显著性差异p《0.01,##表示本发明的塞来昔布乳剂注射液与氟比洛芬酯注射液相比具有显著性差异p《0.05,###表示本发明的塞来昔布乳剂注射液与氟比洛芬酯注射液相比具有显著性差异p《0.01)。因此,本发明的本发明的塞来昔布乳剂注射液具有更好的治疗效果。
[0480]
实施例8:布洛芬浓缩液对角叉菜胶致大鼠足肿胀药效评价
[0481]
市售布洛芬注射剂给药方法为:对于成人的疼痛治疗,每次的给药剂量400mg~800mg,根据需要给药一次,静脉滴注时间必须大于30min。稀释后用于滴注的布洛芬浓度为4mg/ml或更低。每天最大给药剂量为3.2g。
[0482]
参照市售布洛芬注射液对人的给药剂量折算至炎症模型动物(大鼠)的给药剂量,将自制的布洛芬注射液及市售布洛芬注射液配制成对应的给药浓度用于给药。
[0483]
1.实验材料
[0484]
1.1供试品
[0485]
名称:布洛芬注射液
[0486]
配制方法:将0.75g实施例4中浓缩液20(批号2020030501)用25ml 5%葡萄糖注射液稀释至布洛芬的浓度为3.6mg/ml,得到乳剂。所得乳剂的性质如下:
[0487]
布洛芬浓度mdpdi1.05mg/ml38.4nm0.04
[0488]
1.2阳性对照品
[0489]
名称:市售布洛芬注射液caldolor
[0490]
批号:005a19a
[0491]
规格:800mg/支
[0492]
浓度:100mg/ml
[0493]
性状:澄清透明油溶液
[0494]
保存条件及稳定性:25℃避光保存。室温条件下放置2年
[0495]
有效期:09/2025
[0496]
生产厂家:坎伯兰制药(cumberland pharmaceuticals)
[0497]
阳性对照品配制:将市售布洛芬注射液caldolor用5%葡萄糖注射液稀释至布洛芬的浓度为3.6mg/ml。
[0498]
1.3阴性对照
[0499]
名称:5%葡萄糖注射液
[0500]
阴性对照配制:无需配制,直接使用。
[0501]
1.4实验动物
[0502]
品种:sd大鼠
[0503]
级别:spf级动物
[0504]
性别和数量:雄性,30只
[0505]
动物年龄:约6~8周龄
[0506]
动物体重:160~180g
[0507]
动物来源:北京华阜康生物科技股份有限公司
[0508]
2.实验设计
[0509]
2.1sd大鼠足趾肿胀模型的建立及给药
[0510]
动物分组后测定足趾容积,给予相对应药物,30min后多点注射100微升浓度为10mg/ml角叉菜胶溶液,20min后连续测定足趾容积至8小时。
[0511]
2.2剂量和分组
[0512]
给药剂量:人用剂量(400mg/次/70kg),折合到大鼠为36mg/kg
[0513]
分组:将sd大鼠随机分为3组,每组10只。三组分别为市售制剂组(a组,阳性对照,给药浓度为3.6mg/ml)、布洛芬注射液组(b组,将实施例4的浓缩液20用5%葡萄糖注射液稀释至给药浓度为3.6mg/ml);和阴性对照组(c组,5%葡萄糖注射液)。给药方式为尾静脉注射,给药1次。给药细节参见表16。
[0514]
表16.给药方案
[0515][0516]
2.3给药方案
[0517]
尾静脉注射给药,无恢复期。
[0518]
2.4检测指标频率及方法
[0519]
(1)药效学及药动学指标采集
[0520]
分别于注射角叉菜胶后20min、40min、1h、2h、3h、4h、5h、6h、8h测定足趾体积。
[0521]
实验结束后取足部组织匀浆,用于分析测试。
[0522]
3.实验结果
[0523]
3.1一般观察
[0524]
大鼠注射角叉菜胶后,足部肿胀,活动量减少。
[0525]
3.2肿胀体积差值变化
[0526]
各组的足肿胀差值变化如表17所示。
[0527]
表17各组在不同时间点的足肿胀体积差值
[0528][0529]
*与c组(阴性对照组)比较p《0.05;**与c组(阴性对照组)比较p《0.01,a组与b组之间没有显著性差异。
[0530]
3.3肿胀率变化
[0531]
表18各组在不同时间点的足肿胀率
[0532][0533]
*与a组(市售布洛芬注射剂组)比较p《0.05;**与a组(市售布洛芬注射剂组)比较p
《0.01
[0534]
4.结论
[0535]
阳性对照与布洛芬注射液组给药后均有抑制角叉菜胶所致大鼠足肿胀的作用,布洛芬注射液组在给药后40min至8小时与阴性对照组比较具有显著的抑制足肿胀效果,与市售的布洛芬注射剂组抗炎效果相当。
[0536]
通过引用将本文所列出的所有专利和非专利文献的全部内容合并入本文,就如同将它们各自的全部内容逐一列出一样。
[0537]
尽管本文提供了具体实施方案和实施例以对本发明进行举例说明,但是这并不是对本发明范围的限制。基于本公开内容,本领域技术人员能在不背离本发明的精神实质的情况下显而易见地获得其它变型或等同方案,这些变型和等同方案均在本发明的范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1