一种提高宏基因组纳米孔测序数据菌种组装效率的方法与流程
1.本发明涉及生信分析领域,具体涉及一种通过降维提高宏基因组纳米孔测序数据菌种组装效率的方法。
背景技术:
2.宏基因组学(metagenomics,又称元基因组学)是对微生物在其原始生活场所的基因组学研究。宏基因组学直接从环境样品中提取全部微生物的dna或rna,构建宏基因组文库并测序,系统分析该环境中微生物的遗传多样性和功能多样性,以探索分类学、功能和进化等领域。宏基因组学允许我们越过可培养性和分类学特性的限制,直接调查细菌,病毒和真菌等微生物群落的遗传组成。宏基因组学的分析内容主要包括微生物群落的物种组分与差异分析、功能组分与差异分析、以及环境因子与微生物组的关系等。
3.纳米孔测序技术(又称第四代测序技术)是最近几年兴起的新一代测序技术。目前测序长度可以达到150kb。这项技术开始于90年代,经历了三个主要的技术革新:一、单分子dna从纳米孔通过;二、纳米孔上的酶对于测序分子在单核苷酸精度的控制;三、单核苷酸的测序精度控制。目前市场上广泛接受的纳米孔测序平台是oxford nanopore technologies(以下简称ont)公司的minion和gridion纳米孔测序仪。它的特点是单分子测序,有测序读长长,文库制备方便,测序速度快,测序数据实时获取等特点。
4.基于纳米孔测序宏基因组学研究对象是整个生境中的总dna,为了获取环境样品中完整基因组的信息,需要复原每个微生物的全长基因组序列,显然这是理想情况。但是利用宏基因组从头组装技术,即宏基因组reads首先组装成contigs,通过与参考基因组的序列比对,将分类或系统发育信息归于每个contig,得到微生物群落的物种组分,进而进行群落的差异分析,功能分析等。
5.目前纳米孔测序数据的组装分析流程如下:
6.1)在测序运行过程中,使用ont minknow软件收集原始测序数据;
7.2)使用ont albacore或ont guppy软件对原始数据进行碱基序列生成;
8.3)使用自编python脚本过滤掉长度小于500bp和平均测序质量值小于8的序列;
9.4)使用consent软件,进行序列自矫正;
10.5)使用medaka软件进行序列polish;
11.6)使用canu/meta
‑
flye软件进行菌种组装。
12.然而实践中测序reads数据量具大,组装运行时间长,reads利用率低。具体来说由于宏基因组测序针对的是复杂环境下的所有微生物序列,由于物种的多样性和近缘物种的高序列相似性,会给组装增加难度,进而增加组装运行时间。
13.有鉴于此,特提出本发明。
技术实现要素:
14.本发明的目的是寻求提高宏基因组纳米孔测序数据菌种组装效率。为实现上述目
的,本发明提供一种全新思路,在序列组装前通过降维聚类预分群的方式进行测序数据鉴定。
15.具体技术方案如下:
16.本发明首先提供一种通过降维聚类分群提高宏基因组测序数据菌种组装效率的方法,其特征在于,包括如下步骤:
17.步骤1)序列生成:宏基因组测序下机数据生成fastq格式序列信息;
18.步骤2)样本拆分:根据文库标签序列进行样本拆分;
19.步骤3)序列质控:包括但不限于序列长度和/或质量的质控;
20.步骤4)k
‑
mer频率或频数矩阵计算:基于序列进行进行k
‑
mer频率或频数矩阵计算;
21.步骤5)降维聚类分群处理:基于频率或频数矩阵对所有测序序列进行降维聚类分群处理;
22.步骤6)序列组装:降维聚类后分群的每个cluster的序列分别组装。
23.进一步的,所述步骤2)样本拆分为:根据文库的标签序列(比如barcode)将序列拆分成属于不同样本的序列集合,同时还可包括去除接头序列。
24.进一步的,所述步骤3)序列质控为:统计序列的长度和质量值;
25.在一些实施方式中,比如对于纳米孔长读长数据,过滤掉长度小于500bp和平均测序质量值小于8的序列。
26.进一步的,所述步骤4)所述k=2~20000,优选的,所述k=5
‑
75;更优选的,所述k=5,具体的:5
‑
mer的序列种类数为4*4*4*4*4/2=512种,计算每种reads中512种mer的频率或频数,得到5
‑
mer频率或频数矩阵。
27.进一步的,所述步骤5)的降维聚类使用包括但不限于:umap、t
‑
sne、knn进行序列降维聚类;
28.在一些实施方式中,使用umap包进行序列降维聚类;降维聚类的参数设置如下:random_state=42,n_neighbors=30,min_dist=0.0,n_components=2;随后使用python的hdbscan包依据umap降维聚类的结果进行聚类分群并给每条read确定归属于某个cluster。
29.进一步的,所述步骤5)降维聚类分群后的reads序列还可以包括分别进一步做polish处理;优选的,对于每个cluster的reads,使用medaka软件分别进行polish处理。
30.进一步的,所述步骤6)中组装为对每个做过polish的分群cluster的reads分别进行组装;
31.在一些实施方式中,所述组装使用包括但不限于:canu/meta
‑
flye、wtdbg2、necat软件进行。
32.本发明还提供一种物种鉴定的生信分析方法,其特征在于,所述方法包括上述方法,并进一步包括:步骤9)物种鉴定:基于组装后的序列进行物种鉴定。
33.本发明还提供一种物种鉴定的生信分析装置,包括:至少一个存储器,用于存储程序;至少一个处理器,用于加载所述程序以执行如上所述方法。
34.本发明还提供一种存储介质,其中存储有处理器可执行的指令,所述处理器可执行的指令在由处理器执行时用于实现如上述方法。
35.进一步的,上述测序数据为一代、二代、三代或四代测序数据;优选的,为四代纳米孔测序数据。
36.本发明有益的技术效果:
37.本发明通过降维聚类分群把宏基因组数据按照菌种分到不同的cluster里,然后再对每个cluster分别组装,能够显著提高宏基因组组装效率,组装时间至少减少一半以上,与不分群组装的物种鉴定结果一致。
38.本发明有效提高了宏基因组的鉴定效率,同时保证菌种鉴定的有效性和准确性。
附图说明
39.为了更清楚地说明本发明具体实施方式或现有技术中的技术方案,下面将对具体实施方式或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图是本发明的一些实施方式,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
40.图1本发明方法的流程图;
41.图2实施例2中1h的umap降维聚类分群结果图;在四代纳米孔测序平台测得1h产出数据根据本发明中umap降维聚类得到的分群结果,其中每个聚在一起的点是同一个分群;
42.图3实施例2中2h的umap降维聚类分群结果图;在四代纳米孔测序平台测得2h产出数据根据本发明中umap降维得到的分群结果,其中每个聚在一起的点是同一个分群;
43.图4实施例2中3h频率umap降维聚类分群结果图;在四代纳米孔测序平台测得3h产出数据根据本发明中umap降维聚类得到的分群结果,其中每个聚在一起的点是同一个分群;
44.图5实施例2中4h的umap降维聚类分群结果图;在四代纳米孔测序平台测得4h产出数据根据本发明中umap降维聚类得到的分群结果,其中每个聚在一起的点是同一个分群;
45.图6实施例2中5h的umap降维聚类分群结果图;在四代纳米孔测序平台测得5h产出数据根据本发明中umap降维聚类得到的分群结果,其中每个聚在一起的点是同一个分群。
具体实施方式
46.下面将结合实施例对本发明的实施方案进行详细描述,但是本领域技术人员将会理解,下列实施例仅用于说明本发明,而不应视为限制本发明的范围,并且所述实施例是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
47.部分术语定义
48.除非在下文中另有定义,本发明具体实施方式中所用的所有技术术语和科学术语的含义意图与本领域技术人员通常所理解的相同。虽然相信以下术语对于本领域技术人员很好理解,但仍然阐述以下定义以更好地解释本发明。
49.如本发明中所使用,术语“包括”、“包含”、“具有”、“含有”或“涉及”为包含性的(inclusive)或开放式的,且不排除其它未列举的元素或方法步骤。术语“由
…
组成”被认为是术语“包含”的优选实施方案。如果在下文中某一组被定义为包含至少一定数目的实施方案,这也应被理解为揭示了一个优选地仅由这些实施方案组成的组。
50.本发明中的术语“大约”、“大体”表示本领域技术人员能够理解的仍可保证论及特征的技术效果的准确度区间。该术语通常表示偏离指示数值的
±
10%,优选
±
5%。
51.在提及单数形式名词时使用的不定冠词或定冠词例如“一个”或“一种”,“所述”,包括该名词的复数形式。
52.此外,说明书和权利要求书中的术语第一、第二、第三、(a)、(b)、(c)以及诸如此类,是用于区分相似的元素,不是描述顺序或时间次序必须的。应理解,如此应用的术语在适当的环境下可互换,并且本发明描述的实施方案能以不同于本发明描述或举例说明的其它顺序实施。
53.本发明所述的数据降维聚类是一系列相关的高维变量减少为一系列低维变量,这些低维数据会尽可能地反应原始数据的特征,并将相似特征的数据聚为一类。本发明优选采用umap降维聚类算法,基于测序序列的5
‑
mer频数矩阵降维聚类。所述umap(uniform manifold approximation and projection for dimension reduction,一致的流形逼近和投影以进行降维)是一种降维技术,类似于t
‑
sne,可用于可视化,但也可用于一般的非线性降维聚类。
54.本发明所述的宏基因组学(metagenomics,又称元基因组学)是对微生物在其原始生活场所的基因组学研究。宏基因组学直接从环境样品中提取全部微生物的dna或rna,构建宏基因组文库并测序,系统分析该环境中微生物的遗传多样性和功能多样性,以探索分类学、功能和进化等领域。宏基因组学的分析内容主要包括微生物群落的物种组分与差异分析、功能组分与差异分析、以及环境因子与微生物组的关系等。
55.本发明所述的纳米孔测序技术(又称第四代测序技术)是最近几年兴起的新一代测序技术。目前测序长度可以达到150kb。它的特点是单分子测序,有测序读长长,文库制备方便,测序速度快,测序数据实时获取等特点。
56.本发明的通过降维分群提高宏基因组测序数据菌种组装效率的方法,核心在于基于预分群的方式,大体包括如下步骤:步骤1)序列生成:宏基因组测序下机数据生成fastq格式序列信息;步骤2)样本拆分:根据文库标签序列将序列拆分成属于不同样本的序列集合;步骤3)序列质控:比如序列长度和/或质量的质控等;步骤4)k
‑
mer频率或频数矩阵计算,所述k=2~20000;步骤5)降维聚类分群(cluster)处理:基于频率或频数矩阵对所有测序序列进行降维分群处理;步骤6)序列组装:降维后分群cluster的序列分别组装。
57.在一些方面,所述步骤3)包括比如统计序列的长度和质量值。示例性的,对于纳米孔测序数据而言,过滤掉长度小于500bp和平均测序质量值小于8的序列。本领域根据实际测序数据可以适当选择质控标准。
58.在一些方面,所述步骤3)序列质控后还可以进一步包括序列矫正步骤:将过滤后的序列进行自矫正,矫正测序错误的碱基。
59.在一些方面,所述步骤4)中的k=2~20000,优选k=5
‑
75。
60.可以理解本发明中所述的k
‑
mer为一段生物序列中的长度为k的子序列,对于本发明方法中而言k的取值可以是任一正整数,只要满足可以计算k
‑
mer频率都是允许的,因此k的取值原则上可以是>2的正整数当然,当考虑到实际序列长度的限制,k的优选取值2~20000;更优选的为5~75。
61.在一些具体的实例中,以所述k=5为例,5
‑
mer的序列种类数为4*4*4*4*4/2=512
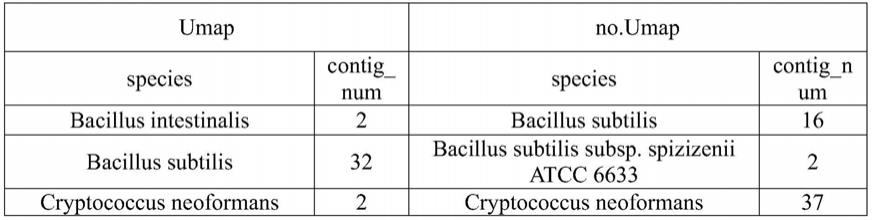
种,计算每种reads中512种mer的频率或频数,得到5
‑
mer频率或频数矩阵。
62.在一些方面,所述步骤5)降维聚类分群后的reads分别进一步做polish处理,比如使用medaka软件分别进行polish处理。
63.在一些具体的实例中,所述步骤5)的降维聚类使用包括但不限于:umap、t
‑
sne、knn进行序列降维聚类;这些不同的降维算法都可以进行聚类操作,并不影响本发明核心。
64.以umap为例,降维的参数设置如下:random_state=42,n_neighbors=30,min_dist=0.0,n_components=2;随后依据umap降维的结果进行聚类分群并给每条read确定归属于某个cluster。
65.在一些方面,所述步骤6)中组装为对每个做过polish的分群cluster的reads分别进行组装;
66.在一些具体的实例中,所述组装使用包括但不限于:canu/meta
‑
flye、wtdbg2、necat软件进行;这些不同的降维聚类算法都可以进行分群聚类,并不影响本发明核心。
67.根据文库的标签序列将序列拆分成属于不同样本的序列集合,同时去除接头序列。
68.可以理解的是本发明的核心思路并不受限于测序平台,理由在于对序列进行k
‑
mer频率或频数的计算并不受测序平台的限制,因此本发明的降维聚类组装方法适用的测序数据包括一代、二代、三代或四代测序数据;优选的,所述测序数据为四代纳米孔测序数据。
69.实施例1本专利方法构建
70.本专利的关注点在于,宏基因组数据预分群后,基于分群后的reads组装提升组装效率。
71.一、方法优化过程
72.首先需要说明两个方面:从reads序列到5
‑
mer频率矩阵,以及每条reads得到的分群cluster标签。
73.在具体计算中,
74.1.首先基于reads序列计算5
‑
mer频率矩阵:
75.‑5‑
mer的序列种类数为4*4*4*4*4/2=512种;
76.‑
计算每种reads中这512种5
‑
mer的频率;
77.‑
得到5
‑
mer频率矩阵;
78.2.然后用umap基于频率矩阵降维,用hdbscan给每条reads分配cluster标签。
79.3.然后用canu/meta
‑
flye软件针对每一个cluster组装。
80.4.最后针对组装结果用blast与nt数据库进行比对,进行物种鉴定。
81.本发明选取了zymobiomics
tm microbial community dna standard(物种已知,为8个细菌和2个真菌)的官方ont的测序数据,按测序时间选取了前5个小时的测序数据,分别为测序1h,2h,3h,4h,5h的下机数据,碱基数据量分别为458m,919m,1.3g,1.7g,2.2g。针对5个时间点的序列来验证降维分群在不同时间点、不同数据量的情况下对组装效率和菌种鉴定的准确性的影响。
82.测试canu软件直接组装全部reads的时间和菌种鉴定结果,与降维聚类分群后用canu分别组装的组装时间和菌种鉴定结果做对比。
83.二、确立本发明分析鉴定流程如下:
84.1.序列生成:ont gridion测序平台产生的数据,通过ont guppy软件将电信号转换为碱基信号,得到fastq格式的序列信息。
85.2.样本拆分:使用ont guppy软件,根据文库的barcode序列将序列拆分成属于不同样本的序列集合,同时去除接头序列。
86.3.序列质控:统计序列的长度和质量值(quality score),对于nanopore长读长数据,过滤掉长度小于500bp或平均测序质量值小于8的序列。
87.4.序列矫正:使用consent软件,将过滤后的序列进行自矫正,矫正测序错误的碱基。
88.5.频率矩阵:使用python脚本计算512种5
‑
mer频率矩阵。
89.6.umap降维:使用python的umap包进行序列降维聚类。参数设置如下:random_state=42,n_neighbors=30,min_dist=0.0,n_components=2。
90.7.hdbscan确定cluster:使用python的hdbscan包依据umap降维的结果给每条read确定归属于某个cluster。
91.8.组装:对于每个cluster的reads,使用canu/meta
‑
flye软件分别进行组装。
92.9.物种鉴定:组装后的contig序列与nt库进行比对,得到物种鉴定结果。
93.实施例2本专利方法umap分群效果
94.本发明通过基于预分群的方式,使zymo官方ont测序数据,在不同时间/数据量梯度下进行分群,来源于相同物种的reads倾向于分到同一个cluster中,具体实施方式基于实施例1的流程进行。
95.umap分群后的降维分群结果见图2
‑
6,图2是1h的降维分群结果图,图3是2h的降维分群结果图,图4是3h的降维分群结果图,图5是4h的降维分群结果图,图6是5h的降维分群结果图。可以看出,通过预分群将全部reads分到不同的cluster中。
96.实施例3本专利方法组装效率评估
97.本发明通过基于预分群的方式,使zymo官方ont测序数据,在不同时间/数据量梯度,如1h~5h的碱基数据量下组装效率有明显提升。具体实施方式基于实施例1的流程进行。
98.组装时间结果表1,可以看出使用umap预分群组装时间缩短接近一半。
99.表1
100.时间base(bp)组装(no_umap)组装时间(umap)1h458,473,60045m47.655s14m36.602s2h919,961,649503m13.250s36m54.974s3h1,375,306,551749m23.833s65m43.655s4h1,796,485,1591126m10.946s154m36.229s5h2,205,881,6981359m9.468s179m0.873s
101.实施例4本专利方法的有效性和准确性
102.本发明用的是zymo的官方数据,此数据中包含的菌种种类已知,所以菌种鉴定结果与zymo菌种进行对比即可验证组装、物种鉴定的准确性。为了验证umap预分群的菌种鉴定的准确性,我们对分群组装后的序列与nt库比对,并与直接组装序列的菌种鉴定结果做
对比。
103.菌种鉴定结果表2(以1h下机数据的结果为例),可以看出菌种鉴定基本一致,并且鉴定出的物种与zymo的物种完全一致,这充分证明了本发明方法的有效性和准确性。
104.表2 1h下机数据物种鉴定结果
105.106.107.[0108][0109]
最后应说明的是:以上各实施例仅用以说明本发明的技术方案,而非对其限制;尽管参照前述各实施例对本发明进行了详细的说明,但本领域的普通技术人员应当理解:其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分或者全部技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本发明各实施例技术方案的范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1