一种用于质谱流式数据挖掘的网络分析方法
1.本发明涉及数据分析模型技术领域,尤其涉及一种用于质谱流式数据挖掘的网络分析方法。
背景技术:
2.系统性红斑狼疮(systemic lupus erythematosus,sle)是一种系统性自身免疫性疾病,可引起皮肤、关节、肾脏和中枢神经系统等多器官损害。疾病的发病模式非常多变,包括皮肤科、肾脏科、神经科、血液科或风湿科,这使得准确诊断和临床治疗面临挑战。尽管存在这些挑战,但对sle发病机制的认识进展有利于临床患者的治疗。
3.系统性红斑狼疮发病机制的研究进展强调了i型干扰素(ifn)和多种免疫细胞类型对自身抗体产生和组织损伤的贡献。慢性ifn的产生及其对免疫系统激活的影响已成为sle的核心机制。ifn是由toll样受体(tlrs)介导的先天性浆细胞树突状细胞(pdcs)激活后产生的。b细胞和t细胞免疫反应的过度激活以及对自身抗原的免疫耐受性的丧失也是sle发病的原因之一。t滤泡辅助细胞可以促进产生自身抗体的b细胞分化。调节性t(treg)细胞可以抑制免疫反应和维持免疫系统,但在sle中细胞的丰度下降。尽管累积性的研究已经为初步理解疾病奠定了基础,但由于免疫系统的细胞复杂性和功能多样性,有必要采用高维度的单细胞技术对系统性的sle发病机制进行研究。
4.质谱流式细胞技术(cytometry by time
‑
of
‑
flight,cytof)极大地增加了单细胞分辨率下同时检测的细胞参数的数量,从而揭示了系统性红斑狼疮发病的细胞和分子基础。结合accense、flowsom、x
‑
shift、monocle 2和phenograph等专业方法,cytof分析发现了tlrs诱导的细胞类型内特异性反应的多样性,以及临床上异质性患者所共有的独特的cd14
hi
单核细胞分泌的细胞因子特征。这些研究说明了cytof在深入分析免疫细胞表型方面的优势,并为系统性红斑狼疮机制提供了宝贵的见解。
5.值得注意的是,cytof的多维性和复杂性还包含着对细胞簇之间"da网络"的高维度洞察的全面认识。洞察多个细胞簇之间的交互模式的异常,可以推进对疾病病因、进展和发病机制相关特征的系统性识别和理解。研究表明,pd1
hi
cxcr5
‑
外周辅助性t细胞在自身免疫性疾病中可以促进b细胞反应和抗体的产生。与识别单个失调的细胞子集相比,了解多个细胞簇之间交互模式的失调,可以提高对疾病相关特征的系统性识别和理解。
6.然而,目前的高维数据分析方法并不能从这类数据集中挖掘出隐藏在细胞簇间的交互作用模式。虽然细胞簇之间的统计相关性和显著性可以很容易地实现,但挖掘高维单细胞蛋白质组学数据的关键挑战是构建和量化跨细胞簇的全局交互网络,以理解sle等多因素复杂型疾病的发病机制。
7.因此,本领域的技术人员致力于开发一种用于质谱流式数据挖掘的网络分析方法。scancell分析方法,将隐藏在高维质谱流式数据中的可用信息转化为细胞簇间的da网络;da网络可以直观地反映免疫系统功能的正常或受损;可以从网络中挖掘出健康样本和疾病样本之间的异常交互模式,有利于疾病发病机制的描述。
技术实现要素:
8.鉴于现有技术的上述缺陷,本发明所要解决的技术问题是scancell分析方法,将隐藏在高维质谱流式数据中的可用信息转化为细胞簇之间的da网络的拓扑结构;通过考虑细胞的蛋白表达量和细胞簇的丰度,为疾病的发病机制提供新的理解维度;挖掘高维蛋白质组数据,理解疾病特异性的细胞和分子畸变。
9.为实现上述目的,本发明提供了一种用于质谱流式数据挖掘的网络分析方法scancell,包括样本分组和细胞类型的da网络的构建。
10.进一步地,所述scancell将所有样本的细胞划分为不同的细胞簇,同时提取簇中相应的细胞丰度直方图作为每个样本的特征。
11.进一步地,所述scancell利用emd计算所有样本两两之间的距离矩阵,将所有样本分为不同的子组,并为每个子组构建一个所有细胞簇的da网络。
12.进一步地,所述scancell利用部分互信息量化细胞簇间的交互作用强度。
13.进一步地,所述da网络,每个节点代表一个细胞簇,每条边表示两个细胞簇之间的交互作用,边的权重表示交互强度。
14.进一步地,所述da网络由cytoscape软件可视化。
15.进一步地,包括如下步骤:
16.步骤1、细胞聚类;
17.步骤2、样本分组;
18.步骤3、da网络的构建。
19.进一步地,所述步骤1包括如下步骤:
20.步骤1.1、所有的sle数据都被一个反双曲正弦(arcsinh)函数标准化;
21.步骤1.2、采用完全无监督的monocle 2算法来完成细胞聚类,从每个样本中选取一定数量的细胞,并将这些选取的细胞合并成一个整体的细胞数据;
22.步骤1.3、应用monocle 2算法将聚合后的细胞进一步划分为若干个细胞簇,计算所有剩余细胞与每个细胞簇中心点之间的距离,将剩余细胞分配到距离最近的细胞簇中。
23.进一步地,所述步骤2包括如下步骤:
24.步骤2.1、从高维数据结构中提取每个样本的有效特征,并将每个样本重新描述为可计算、可操作的数据;
25.步骤2.2、利用emd计算所有样本两两之间的距离矩阵,然后以这个矩阵为输入,利用层次聚类算法对样本进行分组。
26.进一步地,所述步骤3包括如下步骤:
27.步骤3.1、量化两个细胞簇之间的直接关联,并构建所有簇
‑
簇交互作用的全局da网络;
28.步骤3.2、所述da网络保留细胞簇之间的直接交互作用,弱化间接交互作用;
29.步骤3.3、描绘发病机理的是da网络中的异常交互模式,而不是简单的某一细胞簇的异常表达。
30.在本发明的较佳实施方式中,本发明提供一种数据分析模型,特别是涉及高通量质谱流式数据的细胞亚群间的网络分析算法。
31.本发明开发了scancell分析方法,可以将隐藏在高维质谱流式数据中的可用信息
转化为细胞簇之间的da网络的拓扑结构。da网络表征了免疫细胞簇间直接交互作用的强度,弱化了间接交互的干扰,这使得scancell能够在da网络拓扑结构层面上定量识别疾病特异性异常交互模式。scancell通过考虑细胞簇的异常交互模式,为疾病的发病机制提供了新的理解角度。利用scancell分析sle在整个疾病谱中细胞簇间的交互作用,本发明发现,与平稳期sle患者相比,活动期sle患者揭示了cd8
+
t细胞的簇间交互作用减轻。本发明提供了一个工具,用于深入挖掘高维蛋白质组数据,以更好地理解疾病特异性细胞和分子畸变。这是首次以细胞簇间交互作用的角度描绘疾病发病机制的探索性研究,可促进潜在药物靶点的识别。
32.将给定的所有样本的细胞
‑
蛋白分子表达矩阵数据作为输入,scancell利用从样本中提取的特征将所有样本分为不同的"子组",并为每个子组构建一个所有细胞簇的da网络。由一个全局非线性指数计算da,用以表征整个免疫系统中细胞簇的交互强度。样本分组是da网络中挖掘异常交互模式的重要步骤,有助于消除非必要的簇间交互对da网络的干扰作用。此外,样本分组使子组内的样本有其突出的代表性特征,如特定细胞群的富集。
33.一般来说,每个样本被形式化为一个由异质细胞簇组成的高维数据结构,每个细胞都有几十个蛋白表达的特征。在进行样本分组之前,关键的一步是要从当前可用的信息中提取出每个样本的有用特征。基于这一事实,scancell首先将所有样本的细胞划分为不同的细胞簇,同时提取簇中相应的细胞丰度直方图作为每个样本的特征。在原理上,scancell利用细胞簇之间的欧氏距离和每个样本的细胞簇丰度,引入emd来测量样本间的距离矩阵,将同组样本分为不同的子组。scancell把每个子组表示为一个细胞簇
‑
蛋白表达水平的矩阵,并构建其对应的da网络。在da网络中,每个节点代表一个细胞簇,每条边表示两个细胞簇之间的交互作用,其中边的权重表示交互强度。为了使构建的网络能够在系统水平上直接反映免疫细胞簇之间的交互作用,scancell利用部分互信息来量化细胞簇间的交互作用强度。部分互信息是一个基于部分独立性的概率概念计算的非线性度量,它不仅像余弦相似度和欧氏距离那样考虑了成对细胞簇之间的依赖性,同时还考虑了它们与其余细胞簇的关联。构建的da网络由cytoscape软件进行可视化。
34.本发明提出了一个网络分析的方法,揭开了异常的簇间交互作用可能和系统性红斑狼疮发病有关,并从簇间da网络的角度解释了白细胞介素2(il
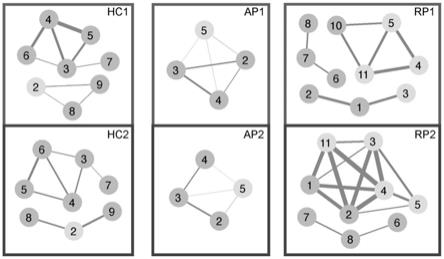
‑
2)的治疗效果。
35.本发明的研究挖掘了系统性红斑狼疮相关的高通量数据中包含的细胞簇间da网络,同时为免疫生物学家深入挖掘疾病中的分子水平上的失调机制提供了一种分析方法。
36.本发明与现有技术相比较,具有如下显而易见的实质性特点和显著优点:
37.1.开发了样本分组和细胞簇之间的直接关联网络,scancell,这是一种新的分析方法,可以将隐藏在高维质谱流式数据中的可用信息转化为细胞簇间的da网络。scancell是第一个构建细胞簇间交互作用的da网络并用于识别疾病特异性的异常特征的方法,这是目前所有现有分析方法所缺乏的。
38.2.虽然没有任何一个细胞簇单独在hcs、aps和rps之间表现出统计学差异,但scancell分析发现aps中cd8
+
t细胞的簇间交互作用较hcs和rps下降,这可能是由于cd8
+
t细胞的效应功能处于过度活跃状态。此外,发现在已被证明可以缓解系统性红斑狼疮的临床症状的白细胞介素2(il
‑
2)的刺激下,可以增强cd8
+
t细胞的簇间交互作用。
39.3.鉴于样本分组保持了子组内样本的同质特征和子组间样本的异质特征,网络构
建弱化了间接交互作用,da网络可以直观地反映免疫系统功能的正常或受损。因此,可以从网络中挖掘出健康样本和疾病样本之间的异常交互模式,有利于揭示疾病发病机制。
40.以下将结合附图对本发明的构思、具体结构及产生的技术效果作进一步说明,以充分地了解本发明的目的、特征和效果。
附图说明
41.图1是本发明的一个较佳实施例的算法挖掘出来的样本整体和样本子组中的细胞亚群交互差异性;
42.图2是本发明的一个较佳实施例的算法挖掘出的不同病人组中细胞亚群交互网络关系的差异。
具体实施方式
43.以下参考说明书附图介绍本发明的多个优选实施例,使其技术内容更加清楚和便于理解。本发明可以通过许多不同形式的实施例来得以体现,本发明的保护范围并非仅限于文中提到的实施例。
44.在附图中,结构相同的部件以相同数字标号表示,各处结构或功能相似的组件以相似数字标号表示。附图所示的每一组件的尺寸和厚度是任意示出的,本发明并没有限定每个组件的尺寸和厚度。为了使图示更清晰,附图中有些地方适当夸大了部件的厚度。
45.一种新型网络分析模型用于质谱流式数据挖掘。
46.步骤1:细胞聚类。
47.1.在细胞聚类之前,所有的sle数据都被一个反双曲正弦(arcsinh)函数标准化。
48.2.这里采用一种完全无监督的monocle 2算法来完成细胞聚类的任务。它首先从每个样本中选取一定数量的细胞,并将这些选取的细胞合并。
49.3.通过应用monocle 2算法将聚合后的细胞进一步划分为若干个细胞簇。然后,通过计算所有剩余细胞与每个细胞簇中心点之间的距离,将剩余细胞分配到距离最近的细胞簇中。也就是说,在scancell中,我们分析的是sle样本的所有细胞,而不仅仅局限于采样后的部分细胞。
50.步骤2:样本分组。
51.1.从高维数据结构中提取每个样本的有效特征,并将每个样本重新描述为可计算、可操作的数据。
52.2.利用emd计算所有样本两两之间的距离矩阵,然后以这个矩阵为输入,利用层次聚类算法对样本进行分组。
53.步骤3:直接交互网络的构建。
54.1.量化两个细胞簇之间的直接关联,并构建所有细胞簇
‑
细胞簇交互作用的da网络。
55.2.这种网络尽可能地保留细胞簇之间的直接交互作用,而弱化间接交互作用,在疾病发病机制分析中发挥了重要作用。
56.3.整个免疫系统的正常功能是靠各种细胞簇的协同作用来维持的,所以网络中的异常交互模式更能准确地反映疾病相关的发病机制,而不是简单的某一细胞簇的异常表
达。
57.如图1所示,活动性系统性红斑狼疮病人cd3+t细胞的da网络。图(左):对没有样本分组的所有活动性狼疮病人(aps)进行细胞簇的da网络构建,图(中):对子组1中的活动性狼疮病人(ap1)进行细胞簇的da网络构建,图(右):对子组2中的活动性狼疮病人(ap2)进行细胞簇的da网络构建。
58.如图2所示,scancell为每个子组构建的da网络。正常组的子组1(hc1,左上),正常组的子组2(hc2,左下);活动性狼疮病人组的子组1(ap1,中上),活动性狼疮病人组的子组2(ap2,中下);稳定性狼疮病人组的子组1(rp1,右上),稳定性狼疮病人组的子组2(rp2,右下)。在da网络中,有编号的圆圈表示细胞簇,边的粗细表示簇间交互作用的强弱。浅颜色的细胞簇表示在两个子组中的交互模式不同。例如,hc1中的cluster 2包含在由cluster 2、cluster 8和cluster 9构建的完全图中,而hc2中的cluster 2则不同。
59.以上详细描述了本发明的较佳具体实施例。应当理解,本领域的普通技术无需创造性改动就可以根据本发明的构思做出诸多修改和变化。因此,凡本技术领域中技术人员依本发明的构思在现有技术的基础上通过逻辑分析、推理或者有限的实验可以得到的技术方案,皆应在由权利要求书所确定的保护范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1