短串联重复序列的致病性分析方法、装置及服务器
1.本技术涉及数据分析领域,具体而言,涉及一种短串联重复序列的致病性分析方法、装置及服务器。
背景技术:
2.发明人发现,wes数据仅仅运用于从中识别短串联重复的异常扩增,但是并未充分挖掘wes数据的潜在价值,探究短串联重复的异常扩增与疾病的关系,造成短串联重复序列致病性分析和解读不规范,以及在分析解读工作中造成大量的人力、物力资源的浪费。
3.针对相关技术中短串联重复序列致病性分析和解读不规范,以及在分析解读工作中造成大量的人力、物力资源的浪费的问题,目前尚未提出有效的解决方案。
技术实现要素:
4.本技术的主要目的在于提供一种短串联重复序列的致病性分析方法、装置及服务器,以解决短串联重复序列致病性分析和解读不规范,以及在分析解读工作中造成大量的人力、物力资源的浪费的问题。
5.为了实现上述目的,根据本技术的一个方面,提供了一种短串联重复序列的致病性分析方法。
6.根据本技术的短串联重复序列的致病性分析方法包括:在wes或wgs数据中筛查出序列集;利用预设分析软件分析所述序列集中的短串联重复序列;根据分析结果从所述序列集中筛除不符合预设筛选条件的短串联重复序列,得到致病序列集。
7.进一步的,在wes或wgs数据中筛查出序列集包括:参考在grch37人类参考基因组上的物理位置、重复序列,分析并筛查出wes/wgs数据中的所有短串联重复序列,作为序列集。
8.进一步的,所述预设分析软件包括:expansion hunter、gangstr、stretch、hipstr、trhist、tredparse、expansion hunter denov中的任意一种。
9.进一步的,利用预设分析软件分析所述序列集中的短串联重复序列包括:利用expansion hunter软件分析所述序列集中的短串联重复序列,得到vcf格式文件和json格式文件;从vcf格式文件中提取短串联重复次数及低质量的短串联重复序列的标记信息。
10.进一步的,根据分析结果从所述序列集中筛除不符合预设筛选条件的短串联重复序列,得到致病序列集包括:根据标记信息从序列集中筛除标记为低质量的短串联重复序列;判断短串联重复次数是否高于预设致病阈值;如果是,则从筛除标记为低质量的短串联重复序列的序列集中筛除高于预设致病阈值的短串联重复序列,得到致病序列集。
11.为了实现上述目的,根据本技术的另一方面,提供了一种短串联重复序列的致病性分析装置。
12.根据本技术的短串联重复序列的致病性分析装置包括:数据筛查模块,用于在wes或wgs数据中筛查出序列集;序列分析模块,用于利用预设分析软件分析所述序列集中的短
串联重复序列;序列筛除模块,用于根据分析结果从所述序列集中筛除不符合预设筛选条件的短串联重复序列,得到致病序列集。
13.进一步的,所述数据筛查模块包括:参考在grch37人类参考基因组上的物理位置、重复序列,分析并筛查出wes/wgs数据中的所有短串联重复序列,作为序列集。
14.进一步的,所述序列分析模块包括:利用expansion hunter软件分析所述序列集中的短串联重复序列,得到vcf格式文件和json格式文件;从vcf格式文件中提取短串联重复次数及低质量的短串联重复序列的标记信息。
15.进一步的,所述序列筛除模块包括:根据标记信息从序列集中筛除标记为低质量的短串联重复序列;判断短串联重复次数是否高于预设致病阈值;如果是,则从筛除标记为低质量的短串联重复序列的序列集中筛除高于预设致病阈值的短串联重复序列,得到致病序列集。
16.为了实现上述目的,根据本技术的另一方面,提供了一种服务器。
17.根据本技术的服务器,包括:存储器和处理器,所述存储器中存储有计算机程序,其中,所述处理器被设置为运行所述计算机程序以执行上述任一项所述的积分展示方法。
18.在本技术实施例中,采用分析短串联重复序列的致病性的方式,通过在wes 或wgs数据中筛查出序列集;利用预设分析软件分析所述序列集中的短串联重复序列;根据分析结果从所述序列集中筛除不符合预设筛选条件的短串联重复序列,得到致病序列集;达到了能够充分挖掘wes数据的潜在价值,探究短串联重复的异常扩增与疾病的关系的目的,从而实现了规范、快速、便捷的对短串联重复序列致病性进行细致的分析和解读,且有效减少在分析解读工作中人力、物力资源浪费的技术效果,进而解决了短串联重复序列致病性分析和解读不规范,以及在分析解读工作中造成大量的人力、物力资源的浪费的技术问题。
附图说明
19.构成本技术的一部分的附图用来提供对本技术的进一步理解,使得本技术的其它特征、目的和优点变得更明显。本技术的示意性实施例附图及其说明用于解释本技术,并不构成对本技术的不当限定。在附图中:
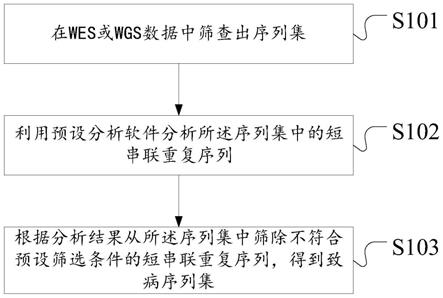
20.图1是根据本技术实施例的短串联重复序列的致病性分析方法的流程示意图;
21.图2是根据本技术实施例的短串联重复序列的致病性分析装置的结构示意图。
具体实施方式
22.为了使本技术领域的人员更好地理解本技术方案,下面将结合本技术实施例中的附图,对本技术实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本技术一部分的实施例,而不是全部的实施例。基于本技术中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都应当属于本技术保护的范围。
23.需要说明的是,本技术的说明书和权利要求书及上述附图中的术语“第一”、“第二”等是用于区别类似的对象,而不必用于描述特定的顺序或先后次序。应该理解这样使用的数据在适当情况下可以互换,以便这里描述的本技术的实施例。此外,术语“包括”和“具有”以及他们的任何变形,意图在于覆盖不排他的包含,例如,包含了一系列步骤或单元的
过程、方法、系统、产品或设备不必限于清楚地列出的那些步骤或单元,而是可包括没有清楚地列出的或对于这些过程、方法、产品或设备固有的其它步骤或单元。
24.在本技术中,术语“上”、“下”、“左”、“右”、“前”、“后”、“顶”、“底”、“内”、“外”、“中”、“竖直”、“水平”、“横向”、“纵向”等指示的方位或位置关系为基于附图所示的方位或位置关系。这些术语主要是为了更好地描述本发明及其实施例,并非用于限定所指示的装置、元件或组成部分必须具有特定方位,或以特定方位进行构造和操作。
25.并且,上述部分术语除了可以用于表示方位或位置关系以外,还可能用于表示其他含义,例如术语“上”在某些情况下也可能用于表示某种依附关系或连接关系。对于本领域普通技术人员而言,可以根据具体情况理解这些术语在本发明中的具体含义。
26.此外,术语“安装”、“设置”、“设有”、“连接”、“相连”、“套接”应做广义理解。例如,可以是固定连接,可拆卸连接,或整体式构造;可以是机械连接,或电连接;可以是直接相连,或者是通过中间媒介间接相连,又或者是两个装置、元件或组成部分之间内部的连通。对于本领域普通技术人员而言,可以根据具体情况理解上述术语在本发明中的具体含义。
27.需要说明的是,在不冲突的情况下,本技术中的实施例及实施例中的特征可以相互组合。下面将参考附图并结合实施例来详细说明本技术。
28.根据本发明实施例,提供了一种短串联重复序列的致病性分析方法,如图1 所示,该方法包括如下的步骤s101至步骤s103:
29.步骤s101、在wes或wgs数据中筛查出序列集;
30.wes数据是指高通量测序中的全外显子测序数据;wgs数据是指全基因组测序数据。以wes数据和wgs数据为基础,从中筛查出符合要求的所有短串联重复序列,作为序列集。
31.根据本发明实施例,优选的,在wes或wgs数据中筛查出序列集包括:
32.参考在grch37人类参考基因组上的物理位置、重复序列,分析并筛查出 wes/wgs数据中的所有短串联重复序列,作为序列集。
33.具体地,由于wes测序具有测序读长短及靶向捕获测序的特点,所以只纳入在wes测序靶向捕获区域范围内的作为目标短串联重复。此外,鉴于wes测序靶向捕获过程中实际捕获到的范围会大于其目标捕获区间,所以针对wes测序数据所采用的捕获区间文件agilent v6,将文件中靶向捕获区间前后100bp的区间都算作是wes测序的有效捕获范围。为了收集到处于wes测序有效捕获范围内的致病短串联重复,首先利用python对agilent v6的bed格式文件中的捕获区间进行调整,在捕获区间原有基础上向前和向后同时扩大100bp得到新的有效捕获区间,然后利用bedtools中的bedtools intersect-a-b命令对收集到的短串联重复和wes测序的有效捕获区间在染色体上的物理位置取交集,得到位于wes测序有效捕获范围内的短串联重复序列,作为序列集。
34.实现了wes或wgs数据的筛查,从中精确的筛查出符合后续分析要求的短串联重复,从而基于这些短串联重复进行致病性分析,可以有效提升致病性分析的精确性。
35.步骤s102、利用预设分析软件分析所述序列集中的短串联重复序列;
36.序列集中包含若干短串联重复;利用预设分析软件进行逐个分析,每个短串联重复均能够得到一分析结果;为后续基于分析结果进行致病性分析提供保障。
37.在本发明实施例中,所述预设分析软件包括:expansion hunter、gangstr、
stretch、hipstr、trhist、tredparse、expansion hunter denov中的任意一种。优选为采用expansion hunter进行短串联重复序列的分析。
38.根据本发明实施例,优选的,利用预设分析软件分析所述序列集中的短串联重复序列包括:
39.利用expansion hunter软件分析所述序列集中的短串联重复序列,得到vcf 格式文件和json格式文件;
40.从vcf格式文件中提取短串联重复次数及低质量的短串联重复序列的标记信息。
41.expansion hunter是一款基于二代测序数据预测短串联重复长度的软件。我们采用expansion hunter来预测已知致病短串联重复在样本中的重复情况。
42.具体地,运行该软件需要的输入文件包含以下3个部分:(1)利用python将已知致病短串联重复生成符合要求的json文件,(2)已经比对到grch37参考基因组上的wes测序样本的bam文件及其的索引文件,(3)比对wes测序文件到grch37 参考基因组上的过程中所使用的对应版本的fasta文件。准备好输入文件后按照以下代码运行expansion hunter得到对应的结果文件。
43.expansion hunter软件运行代码如下:
[0044][0045]
expansion hunter软件运行完成后,每一个wes测序的样本都会得到对应的结果文件。expansion hunter软件的结果文件包括一个vcf格式文件和一个json 格式文件。其中vcf格式文件里面的内容包含了短串联重复的基因型和位置信息, vcf文件中的每一行为一条短串联重复的详细信息其中包括该短串联重复在一对同源染色体上的重复次数、该短串联重复在参考基因组上的重复次数、测序时该区域所测到的平均深度等信息。json格式文件的内容在vcf格式文件的基础上多出了expansion hunter软件运行过程中所使用到bam文件中测序读段的相关信息如短串联重复涉及到几条测序读段、测序读段的平均长度等。鉴于判断短串联重复是否致病的关键指标是其重复次数,所以只针对expansion hunter软件结果中的vcf格式文件进行了后续处理。
[0046]
经过处理得到vcf格式文件中的短串联重复次数及低质量的短串联重复序列的标记信息;基于这两个数据可以为致病性的短串联重复序列的筛选提供保障。
[0047]
步骤s103、根据分析结果从所述序列集中筛除不符合预设筛选条件的短串联重复序列,得到致病序列集。
[0048]
判断分析结果中的信息是否符合预设筛选条件,如果不符合就筛除不符合的短串联重复,如果符合就不执行任何操作,将该符合的短串联重复保留在序列集中;对每个短串联重复均执行以上的判断,最终不符合筛选条件的全部被筛除,剩余的所有短串联重复即为致病序列集。能够充分挖掘wes数据的潜在价值,探究短串联重复的异常扩增与疾病的关
系,实现了规范、快速、便捷的对短串联重复序列致病性进行细致的分析和解读,且有效减少在分析解读工作中人力、物力资源浪费。
[0049]
根据本发明实施例,优选的,根据分析结果从所述序列集中筛除不符合预设筛选条件的短串联重复序列,得到致病序列集包括:
[0050]
根据标记信息从序列集中筛除标记为低质量的短串联重复序列;
[0051]
判断短串联重复次数是否高于预设致病阈值;
[0052]
如果是,则从筛除标记为低质量的短串联重复序列的序列集中筛除高于预设致病阈值的短串联重复序列,得到致病序列集。
[0053]
具体地,分析结果中包含标记信息和短串联重复次数,根据标记信息判断短串联重复序列是否符合标记为低质量的短串联重复序列,如果是,则筛除标记为低质量的短串联重复序列;再根据短串联重复次数判断其是否高于预设致病阈值,如果是,则进一步筛除高于预设致病阈值的短串联重复序列,最终序列集中剩余的短串联重复即为致病序列集。通过两次筛选,提高了最终致病的短串联重复序列的正确度,大大降低了误判几率。
[0054]
从以上的描述中,可以看出,本发明实现了如下技术效果:
[0055]
在本技术实施例中,采用分析短串联重复序列的致病性的方式,通过在wes 或wgs数据中筛查出序列集;利用预设分析软件分析所述序列集中的短串联重复序列;根据分析结果从所述序列集中筛除不符合预设筛选条件的短串联重复序列,得到致病序列集;达到了能够充分挖掘wes数据的潜在价值,探究短串联重复的异常扩增与疾病的关系的目的,从而实现了规范、快速、便捷的对短串联重复序列致病性进行细致的分析和解读,且有效减少在分析解读工作中人力、物力资源浪费的技术效果,进而解决了短串联重复序列致病性分析和解读不规范,以及在分析解读工作中造成大量的人力、物力资源的浪费的技术问题。
[0056]
需要说明的是,在附图的流程图示出的步骤可以在诸如一组计算机可执行指令的计算机系统中执行,并且,虽然在流程图中示出了逻辑顺序,但是在某些情况下,可以以不同于此处的顺序执行所示出或描述的步骤。
[0057]
根据本发明实施例,还提供了一种用于实施上述短串联重复序列的致病性分析方法的装置,如图2所示,该装置包括:
[0058]
数据筛查模块10,用于在wes或wgs数据中筛查出序列集;
[0059]
wes数据是指高通量测序中的全外显子测序数据;wgs数据是指全基因组测序数据。以wes数据和wgs数据为基础,从中筛查出符合要求的所有短串联重复序列,作为序列集。
[0060]
优选的,所述数据筛查模块10包括:
[0061]
参考在grch37人类参考基因组上的物理位置、重复序列,分析并筛查出 wes/wgs数据中的所有短串联重复序列,作为序列集。
[0062]
具体地,由于wes测序具有测序读长短及靶向捕获测序的特点,所以只纳入在wes测序靶向捕获区域范围内的作为目标短串联重复。此外,鉴于wes测序靶向捕获过程中实际捕获到的范围会大于其目标捕获区间,所以针对wes测序数据所采用的捕获区间文件agilent v6,将文件中靶向捕获区间前后100bp的区间都算作是wes测序的有效捕获范围。为了收集到处于wes测序有效捕获范围内的致病短串联重复,首先利用python对agilent v6的bed格式文件中的捕获区间进行调整,在捕获区间原有基础上向前和向后同时扩大
100bp得到新的有效捕获区间,然后利用bedtools中的bedtools intersect-a-b命令对收集到的短串联重复和wes测序的有效捕获区间在染色体上的物理位置取交集,得到位于wes测序有效捕获范围内的短串联重复序列,作为序列集。
[0063]
实现了wes或wgs数据的筛查,从中精确的筛查出符合后续分析要求的短串联重复,从而基于这些短串联重复进行致病性分析,可以有效提升致病性分析的精确性。
[0064]
序列分析模块20,用于利用预设分析软件分析所述序列集中的短串联重复序列;
[0065]
序列集中包含若干短串联重复;利用预设分析软件进行逐个分析,每个短串联重复均能够得到一分析结果;为后续基于分析结果进行致病性分析提供保障。
[0066]
在本发明实施例中,所述预设分析软件包括:expansion hunter、gangstr、stretch、hipstr、trhist、tredparse、expansion hunter denov中的任意一种。优选为采用expansion hunter进行短串联重复序列的分析。
[0067]
优选的,所述序列分析模块20包括:
[0068]
利用expansion hunter软件分析所述序列集中的短串联重复序列,得到vcf 格式文件和json格式文件;
[0069]
从vcf格式文件中提取短串联重复次数及低质量的短串联重复序列的标记信息。
[0070]
expansion hunter是一款基于二代测序数据预测短串联重复长度的软件。我们采用expansion hunter来预测已知致病短串联重复在样本中的重复情况。
[0071]
具体地,运行该软件需要的输入文件包含以下3个部分:(1)利用python将已知致病短串联重复生成符合要求的json文件,(2)已经比对到grch37参考基因组上的wes测序样本的bam文件及其的索引文件,(3)比对wes测序文件到grch37 参考基因组上的过程中所使用的对应版本的fasta文件。准备好输入文件后按照以下代码运行expansion hunter得到对应的结果文件。
[0072]
expansion hunter软件运行代码如下:
[0073][0074]
expansion hunter软件运行完成后,每一个wes测序的样本都会得到对应的结果文件。expansion hunter软件的结果文件包括一个vcf格式文件和一个json 格式文件。其中vcf格式文件里面的内容包含了短串联重复的基因型和位置信息, vcf文件中的每一行为一条短串联重复的详细信息其中包括该短串联重复在一对同源染色体上的重复次数、该短串联重复在参考基因组上的重复次数、测序时该区域所测到的平均深度等信息。json格式文件的内容在vcf格式文件的基础上多出了expansion hunter软件运行过程中所使用到bam文件中测序读段的相关信息如短串联重复涉及到几条测序读段、测序读段的平均长度等。鉴于判断短串联重复是否致病的关键指标是其重复次数,所以只针对expansion hunter软件结果中的vcf格式文件进行了后续处理。
[0075]
经过处理得到vcf格式文件中的短串联重复次数及低质量的短串联重复序列的标记信息;基于这两个数据可以为致病性的短串联重复序列的筛选提供保障。
[0076]
序列筛除模块30,用于根据分析结果从所述序列集中筛除不符合预设筛选条件的短串联重复序列,得到致病序列集。
[0077]
判断分析结果中的信息是否符合预设筛选条件,如果不符合就筛除不符合的短串联重复,如果符合就不执行任何操作,将该符合的短串联重复保留在序列集中;对每个短串联重复均执行以上的判断,最终不符合筛选条件的全部被筛除,剩余的所有短串联重复即为致病序列集。能够充分挖掘wes数据的潜在价值,探究短串联重复的异常扩增与疾病的关系,实现了规范、快速、便捷的对短串联重复序列致病性进行细致的分析和解读,且有效减少在分析解读工作中人力、物力资源浪费。
[0078]
优选的,所述序列筛除模块30包括:
[0079]
根据标记信息从序列集中筛除标记为低质量的短串联重复序列;
[0080]
判断短串联重复次数是否高于预设致病阈值;
[0081]
如果是,则从筛除标记为低质量的短串联重复序列的序列集中筛除高于预设致病阈值的短串联重复序列,得到致病序列集。
[0082]
具体地,分析结果中包含标记信息和短串联重复次数,根据标记信息判断短串联重复序列是否符合标记为低质量的短串联重复序列,如果是,则筛除标记为低质量的短串联重复序列;再根据短串联重复次数判断其是否高于预设致病阈值,如果是,则进一步筛除高于预设致病阈值的短串联重复序列,最终序列集中剩余的短串联重复即为致病序列集。通过两次筛选,提高了最终致病的短串联重复序列的正确度,大大降低了误判几率。
[0083]
从以上的描述中,可以看出,本发明实现了如下技术效果:
[0084]
在本技术实施例中,采用分析短串联重复序列的致病性的方式,通过在wes 或wgs数据中筛查出序列集;利用预设分析软件分析所述序列集中的短串联重复序列;根据分析结果从所述序列集中筛除不符合预设筛选条件的短串联重复序列,得到致病序列集;达到了能够充分挖掘wes数据的潜在价值,探究短串联重复的异常扩增与疾病的关系的目的,从而实现了规范、快速、便捷的对短串联重复序列致病性进行细致的分析和解读,且有效减少在分析解读工作中人力、物力资源浪费的技术效果,进而解决了短串联重复序列致病性分析和解读不规范,以及在分析解读工作中造成大量的人力、物力资源的浪费的技术问题。
[0085]
显然,本领域的技术人员应该明白,上述的本发明的各模块或各步骤可以用通用的计算装置来实现,它们可以集中在单个的计算装置上,或者分布在多个计算装置所组成的网络上,可选地,它们可以用计算装置可执行的程序代码来实现,从而,可以将它们存储在存储装置中由计算装置来执行,或者将它们分别制作成各个集成电路模块,或者将它们中的多个模块或步骤制作成单个集成电路模块来实现。这样,本发明不限制于任何特定的硬件和软件结合。
[0086]
以上所述仅为本技术的优选实施例而已,并不用于限制本技术,对于本领域的技术人员来说,本技术可以有各种更改和变化。凡在本技术的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本技术的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1