药物组合物及其制备方法和应用与流程
1.本发明涉及药物制剂技术领域,具体而言,涉及一种药物组合物及其制备方法和应用。
背景技术:
2.黄芩苷和野黄芩苷都是黄酮类糖苷(简称黄酮苷),具有丰富的药理活性,例如通过抗脂质过氧化提高抗氧化能力,清除自由基和超氧阴离子作用,改善血液循环增加血流量,抗血小板聚集,抑制病毒感染,增强免疫力,抗细胞缺氧,神经保护,抑制肿瘤细胞生长等。
3.临床应用证明黄酮苷类天然产物的抗菌消炎作用确切,在胃肠道疾病的治疗中具有抗炎、抑菌、解痉等方面的功效。但是,由于黄芩苷水溶性低,市面上流通的大部分普通黄芩苷制剂口服后,由于黄芩苷在胃肠液的溶解度低,透过生物膜进入血循环的量少,所以其口服生物利用度很低,临床中只能通过增大给药剂量才能起到一定治疗效果,但给药剂量的增加势必会给患者的服药和制剂工艺带来诸多不便。
4.因此,如何提高黄酮苷类天然药物的生物利用度以及生物活性是亟待解决的技术问题。
技术实现要素:
5.基于此,有必要提供一种生物利用度更高、生物活性更好的药物组合物及其制备方法和应用。
6.为了实现本发明的上述目的,特采用以下技术方案:
7.本发明提供一种药物组合物,包括纳米黄酮苷和药物活性小分子,
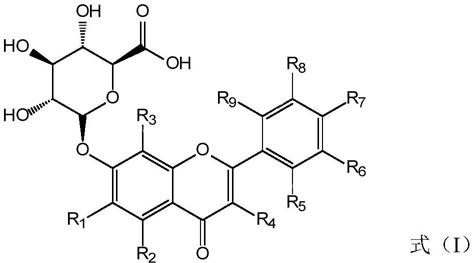
8.所述纳米黄酮苷具有式(i)所示的结构通式:
[0009][0010]
其中,r1~r9各自独立地选自
‑
h、
‑
oh、c1~c6烷基、烷氧基或取代烷基,且r1和r2中至少有一个为
‑
oh;
[0011]
所述药物活性小分子为伊马替尼、伊布替尼、吉非替尼、厄洛替尼、尼洛替尼、达沙替尼、拉帕替尼、培唑帕尼、奥希替尼、帕波西尼、加兰他敏、力帆斯的明、美金刚、芬戈莫德、西格列汀、维格列汀、沙格列汀、阿格列汀、利格列汀、达格列净、恩格列净、卡格列净、奥司他韦、金刚烷胺、羟氯喹、罗红霉素、阿奇霉素、阿莫西林、头孢克洛、头孢克肟、头孢布烯、头
孢他美、头孢呋辛、美罗培南、替比培南、氧氟沙星、左氧氟沙星、环丙沙星、莫西沙星、拉米夫定、替比夫定、替诺福韦、恩替卡韦、阿德福韦、索非布韦、达拉他韦、阿舒瑞韦、齐多夫定、恩曲他滨、司他夫定、奈韦拉平、茚地那韦、利托那韦、达芦那韦、拉替拉韦和多替拉韦中任意一种。。
[0012]
在一些实施方式中,r1和r2均选自
‑
oh。
[0013]
在一些实施方式中,所述纳米黄酮苷为纳米黄芩苷或纳米野黄芩苷。
[0014]
在一些实施方式中,所述纳米黄酮苷和所述药物活性小分子的质量比为1:100至100:1。
[0015]
在一些实施方式中,所述纳米黄酮苷和所述所述药物活性小分子的质量比为1:10至10:1。
[0016]
本发明又一方面,还提供一种所述药物组合物的制备方法,包括以下步骤:
[0017]
将黄酮苷研磨制成纳米黄酮苷,纳米黄酮苷和药物活性小分子混合。
[0018]
在一些实施方式中,所述将黄酮苷研磨制成纳米黄酮苷的步骤中,研磨的转速为1000rpm~3000rpm,研磨的时间为10min~40min。
[0019]
在一些实施方式中,所述药物组合物含有治疗有效量的所述纳米黄酮苷和所述药物活性小分子,以及药学上可接受的载体、赋形剂或稀释剂。
[0020]
进一步,本发明再提供一种所述药物组合物的制备方法,包括以下步骤:
[0021]
将黄酮苷和药物活性小分子混合,混合物经研磨得到组合物。
[0022]
在一些实施方式中,所述混合物经研磨得到组合物的步骤中,研磨的转速为2000rpm~3000rpm,研磨的时间为20min~60min。
[0023]
本发明再一方面,提供所述药物组合物在制备抗炎症药物中的应用,其中,所述纳米黄酮苷为纳米黄芩苷或纳米野黄芩苷,所述药物活性小分子为羟氯喹。
[0024]
在一些实施方式中,所述抗炎症药物用于炎症疾病的治疗,所述炎症疾病为疟疾、类风湿性关节炎、系统性红斑狼疮、斑疹性关节炎、斑块状切口炎或q热等风湿性疾病。
[0025]
与现有技术相比,本发明的有益效果为:
[0026]
本发明提供的纳米黄酮苷和药物活性小分子组合物,纳米黄酮苷相比较于大粒径的黄酮苷,溶解度增加,溶解速度加快,将其与药物活性小分子联合使用,二者可以产生协同作用,相较于二者的单独使用,二者联用具有更高的生物活性和生物利用度。而且将该组合物作为药物制剂后,药物溶出度更好,可以被人体更多吸收并发挥药效作用,为满足临床需求提供了保障。
具体实施方式
[0027]
以下结合具体实施例对本发明进行进一步详细的说明。本发明可以以许多不同的形式来实现,并不限于本发明所描述的实施方式。相反地,提供这些实施方式的目的是使对本发明公开内容理解更加透彻全面。
[0028]
除非另有定义,本发明所使用的所有的技术和科学术语与属于本发明的技术领域的技术人员通常理解的含义相同。本文中在本发明的说明书中所使用的术语只是为了描述具体的实施例的目的,不是旨在于限制本发明。本文所使用的术语“和/或”包括一个或多个相关的所列项目的任意的和所有的组合。
[0029]
术语和定义
[0030]
除非另外说明或存在矛盾之处,本文中使用的术语或短语具有以下含义:
[0031]
术语“烷基”是指包含伯(正)碳原子、或仲碳原子、或叔碳原子、或季碳原子、或其组合的饱和烃。包含该术语的短语,例如,“c1~c6烷基”是指包含1~6个碳原子的烷基,每次出现时,可以互相独立地为c1烷基、c2烷基、c3烷基、c4烷基、c5烷基或c6烷基。合适的实例包括但不限于:甲基(me、
‑
ch3)、乙基(et、
‑
ch2ch3)、1
‑
丙基(n
‑
pr、n
‑
丙基、
‑
ch2ch2ch3)、2
‑
丙基(i
‑
pr、i
‑
丙基、
‑
ch(ch3)2)、1
‑
丁基(n
‑
bu、n
‑
丁基、
‑
ch2ch2ch2ch3)、2
‑
甲基
‑1‑
丙基(i
‑
bu、i
‑
丁基、
‑
ch2ch(ch3)2)、2
‑
丁基(s
‑
bu、s
‑
丁基、
‑
ch(ch3)ch2ch3)、2
‑
甲基
‑2‑
丙基(t
‑
bu、t
‑
丁基、
‑
c(ch3)3)、1
‑
戊基(n
‑
戊基、
‑
ch2ch2ch2ch2ch3)、2
‑
戊基(
‑
ch(ch3)ch2ch2ch3)、3
‑
戊基(
‑
ch(ch2ch3)2)、2
‑
甲基
‑2‑
丁基(
‑
c(ch3)2ch2ch3)、3
‑
甲基
‑2‑
丁基(
‑
ch(ch3)ch(ch3)2)、3
‑
甲基
‑1‑
丁基(
‑
ch2ch2ch(ch3)2)、2
‑
甲基
‑1‑
丁基(
‑
ch2ch(ch3)ch2ch3)、1
‑
己基(
‑
ch2ch2ch2ch2ch2ch3)、2
‑
己基(
‑
ch(ch3)ch2ch2ch2ch3)、3
‑
己基(
‑
ch(ch2ch3)(ch2ch2ch3))、2
‑
甲基
‑2‑
戊基(
‑
c(ch3)2ch2ch2ch3)、3
‑
甲基
‑2‑
戊基(
‑
ch(ch3)ch(ch3)ch2ch3)、4
‑
甲基
‑2‑
戊基(
‑
ch(ch3)ch2ch(ch3)2)、3
‑
甲基
‑3‑
戊基(
‑
c(ch3)(ch2ch3)2)、2
‑
甲基
‑3‑
戊基(
‑
ch(ch2ch3)ch(ch3)2)、2,3
‑
二甲基
‑2‑
丁基(
‑
c(ch3)2ch(ch3)2)、和3,3
‑
二甲基
‑2‑
丁基(
‑
ch(ch3)c(ch3)3。
[0032]
术语“烷氧基”是指具有
‑
o
‑
烷基的基团,即如上所定义的烷基经由氧原子连接至母核结构。包含该术语的短语,例如,“c1~c6烷氧基”是指烷基部分包含1~6个碳原子,每次出现时,可以互相独立地为c1烷氧基、c2烷氧基、c3烷氧基、c4烷氧基、c5烷氧基或c6烷氧基。合适的实例包括但不限于:甲氧基(
‑
o
‑
ch3或
‑
ome)、乙氧基(
‑
o
‑
ch2ch3或
‑
oet)和叔丁氧基(
‑
o
‑
c(ch3)3或
‑
otbu)。
[0033]“药学上可接受的”指在合理医学判断范围内适于施用患者且与合理益处/风险比相称的那些配体、材料、组合物和/或剂型。
[0034]“药学上可接受的载体、赋形剂或稀释剂”指药学上可接受的材料、组合物或媒剂,例如液体或固体填充剂、稀释剂、赋形剂、溶剂或囊封材料。如本文所用,语言“药学上可接受的载体、赋形剂或稀释剂”包括与药物施用相容的缓冲剂、注射用无菌水、溶剂、分散介质、包衣、抗细菌剂及抗真菌剂、等渗剂及吸收延迟剂及诸如此类。在与配制物中其他成分兼容且对患者无害的意义上,每种载体、赋形剂或稀释剂必须为“药学上可接受的”。合适的实例包括但不限于:(1)糖,例如乳糖、葡萄糖及蔗糖;(2)淀粉,例如玉米淀粉、马铃薯淀粉及经取代或未经取代的β
‑
环糊精;(3)纤维素及其衍生物,例如羧甲基纤维素钠、乙基纤维素及乙酸纤维素;(4)粉状黄蓍胶;(5)麦芽;(6)明胶;(7)滑石;(8)赋形剂,例如可可脂及栓剂蜡;(9)油类,例如花生油、棉籽油、红花油、芝麻油、橄榄油、玉米油及大豆油;(10)二醇,例如丙二醇;(11)多元醇,例如甘油、山梨醇、甘露醇及聚乙二醇;(12)酯类,例如油酸乙酯及月桂酸乙酯;(13)琼脂;(14)缓冲剂,例如氢氧化镁及氢氧化铝;(15)海藻酸;(16)无热原水;(17)等渗盐水;(18)林格氏溶液;(19)乙醇;(20)磷酸盐缓冲液;及(21)药物配制物中所采用的其他无毒兼容物质。
[0035]
涉及基团的“取代的”指与基团内的成员原子附接的一个或多个氢原子由选自所限定或适合的取代基中的取代基替代。应理解术语“取代的”包括下述隐含条件:这种取代应与取代的原子和取代基的允许化合价一致并且取代导致稳定的化合物。当陈述基团可以
含有一个或多个取代基时,该基团内的一个或多个成员原子可以被取代。另外,只要这种取代与原子的允许化合价一致,该基团内的单个成员原子就可以由多于一种取代基取代。“成员原子”指的是形成链或环的一个原子或多个原子。在链中以及环内存在多于一个成员原子的情况下,每个成员原子与该链或环中的相邻成员原子共价结合。组成链或环上的取代基的原子不是该链或环中的成员原子。
[0036]
组合物
[0037]
本发明提供一种药物组合物,包括纳米黄酮苷和药物活性小分子。
[0038]
纳米黄酮苷具有式(i)所示的结构通式:
[0039][0040]
其中,r1~r9各自独立地选自
‑
h、
‑
oh、c1~c6烷基、烷氧基或取代烷基,且r1和r2中至少有一个为
‑
oh。
[0041]
在一些实施方式中,r1和r2均选自
‑
oh。
[0042]
在一些实施方式中,r3选自
‑
h或
‑
och3。
[0043]
在一些实施方式中,r5、r6、r9均选自
‑
h。
[0044]
在一些实施方式中,r7、r8各自独立地选自
‑
h或
‑
oh。
[0045]
在一些实施方式中,r8选自
‑
h。
[0046]
在一些实施方式中,r7选自
‑
oh。
[0047]
在另一些实施方式中,r7选自
‑
h。
[0048]
在一些实施方式中,纳米黄酮苷为纳米芹菜素黄酮苷、纳米黄芩苷、纳米野黄芩苷、纳米白杨素黄酮苷或纳米汉黄芩苷。
[0049]
优选的,纳米黄酮苷为纳米黄芩苷或纳米野黄芩苷。
[0050]
纳米黄芩苷的分子结构如式(i
‑
i)所示:
[0051][0052][0053]
所述野黄芩苷的分子结构如式(i
‑
ii)所示:
[0054][0055]
在一些实施方式中,纳米黄酮苷的粒径分布范围为50nm~500nm。该粒径分布范围内的纳米黄酮苷具有更好的溶解度和溶解速度。
[0056]
药物活性小分子的具体实例可以包括但不限于,伊马替尼、伊布替尼、吉非替尼、厄洛替尼、尼洛替尼、达沙替尼、拉帕替尼、培唑帕尼、奥希替尼、帕波西尼、加兰他敏、力帆斯的明、美金刚、芬戈莫德、西格列汀、维格列汀、沙格列汀、阿格列汀、利格列汀、达格列净、恩格列净、卡格列净、奥司他韦、金刚烷胺、羟氯喹、罗红霉素、阿奇霉素、阿莫西林、头孢克洛、头孢克肟、头孢布烯、头孢他美、头孢呋辛、美罗培南、替比培南、氧氟沙星、左氧氟沙星、环丙沙星、莫西沙星、拉米夫定、替比夫定、替诺福韦、恩替卡韦、阿德福韦、索非布韦、达拉他韦、阿舒瑞韦、齐多夫定、恩曲他滨、司他夫定、奈韦拉平、茚地那韦、利托那韦、达芦那韦、拉替拉韦和多替拉韦。
[0057]
在一些实施方式中,纳米黄酮苷和药物活性小分子的质量比为1:100至100:1之间的任意值,例如还可以为1:50、1:25、1:20、1:15、1:10、1:5、1:1、5:1、10:1、15:1、20:1、25:1、50:1等。
[0058]
在一些优选实施方式中,纳米黄酮苷和药物活性小分子的质量比为1:10至10:1之间的任意值。
[0059]
在一些实施方式中,所述药物组合物含有治疗有效量的上述任意实施方式的纳米
黄酮苷和药物活性小分子,以及药学上可接受的载体、赋形剂或稀释剂。
[0060]
本发明还提供一种上述药物组合物的制备方法,包括以下步骤:
[0061]
s10,将黄酮苷研磨制成纳米黄酮苷,纳米黄酮苷和药物活性小分子混合。
[0062]
步骤s10中,未研磨之前的黄酮苷通常为市售未经研磨的粒径大于1微米的黄酮苷。
[0063]
在一些实施方式中,步骤s10中,研磨的转速独立选自1000rpm~3000rpm之间的任意值,研磨的时间独立选自10min~40min之间的任意值。
[0064]
步骤s10可以包括,向黄酮苷中加入助悬剂和水一起研磨,制成黄酮苷纳米混悬液,再向黄酮苷纳米混悬液中加入药物活性小分子,将纳米黄酮苷和药物活性小分子混合。加入助悬剂的目的是为了进一步分散纳米黄酮苷,防止其团聚。
[0065]
所述助悬剂的实例可以包括但不限于,聚乙二醇、羟丙基甲基纤维素、羟丙基纤维素、甲基纤维素、聚乙烯吡咯烷酮、聚乙二醇、脂肪酸甘油酯、多元醇型非离子表面活性剂、聚氧乙烯型非离子表面洁性剂、泊洛沙姆、吐温、维生素e聚乙二醇琥珀酸醋、磷脂、明胶、黄原胶、十二烷基硫酸钠、脱氧胆酸钠以及它们的组合。
[0066]
本发明进一步还提供上述药物组合物的另一种制备方法,包括以下步骤:
[0067]
s30,将黄酮苷和药物活性小分子混合,混合物经研磨得到组合物。
[0068]
步骤s30中,未研磨之前的黄酮苷通常为市售未经研磨的粒径大于1微米的黄酮苷。
[0069]
在一些实施方式中,步骤s40中,研磨的转速独立选自2000rpm~3000rpm之间的任意值,研磨的时间独立选自20min~60min之间的任意值。所述研磨使用的纳米研磨机工作腔直径为85mm。如纳米研磨机工作腔直径有变化,应相应调节转速。
[0070]
步骤s30可以包括,将大粒径黄酮苷、药物活性小分子、助悬剂和水一起研磨,制成纳米混悬液,再将纳米混悬液干燥得到纳米混合物颗粒。步骤s20则为,将纳米颗粒和药物活性小分子混合。加入助悬剂的目的是为了进一步分散黄酮苷,防止其团聚。
[0071]
该步骤中的助悬剂可以与上述助悬剂相同,在此不再赘述。
[0072]
在一些实施例中,所述纳米黄酮苷选自纳米黄芩苷或纳米野黄芩苷,所述药物活性小分子选自羟氯喹。
[0073]
进一步,本发明还涉及上述药物组合物在制备抗炎症药物中的应用,其中,该组合物中纳米黄酮苷选自纳米黄芩苷或纳米野黄芩苷,药物活性小分子选自羟氯喹。
[0074]
在一些实施方式中,所述炎症疾病为疟疾、类风湿性关节炎、系统性红斑狼疮、斑疹性关节炎、斑块状切口炎或q热等风湿性疾病。
[0075]
给药和配制品
[0076]
含有本发明的药物组合物的药物的生产及其应用可以根据熟知的制药方法进行。
[0077]
虽然根据本发明可用于治疗的本发明的组合物可以以原始化学化合物的形式给药,但是优选地是与一种或多种助剂、赋形剂、载体、缓冲剂、稀释剂和/或其他常规药物辅料一起将活性成分在药物组合物中引入。本发明的组合物可以是无水的或溶剂化的。
[0078]
在优选实施方式中,本发明提供药物,其包括根据本发明可用的组合物以及用于其的一种或多种药学上可接受的载体和可选地其他治疗性和/或预防性成分。该一种或多种载体必须是在与配制品的其他成分相容的且对其受体无害的意义上是“可接受的”。
[0079]
本发明的药物可以是适用于口服、直肠、支气管、鼻腔、局部、口腔、舌下、经皮、阴道或肠胃外(包括皮肤、皮下、肌内、腹膜内、静脉内、动脉内、脑内、眼内注射或输注)给药的那些药物,或者为适用于通过吸入或吹气给药(包括粉末和液体气雾剂给药)或通过缓释体系给药的形式的那些药物。缓释体系的适合示例包括含有本发明的组合物的固体疏水聚合物的半渗透基质,该基质可以是成形物品的形式,例如膜或微胶囊。
[0080]
根据本发明可用的组合物与常规助剂、载体或稀释剂一起可以因此被放置成药物及其单位剂量的形式。这样的形式包括:固体,特别地为片剂、填充胶囊、粉剂和药丸(pellet)形式;以及液体,特别地为含水或非水溶液剂、悬浮剂、乳剂、万能药(elixir)和用其装填的胶囊,用于口服的所有形式,用于直肠给药的栓剂以及用于肠胃外使用的无菌注射溶液。这些药物和其单位剂量形式可以在有或没有其他活性化合物或组成部分的情况下包括常规比例的常规成分,且这种单位剂量形式可以含有与待使用的预期日常剂量范围相应的任何适合有效量的活性成分。
[0081]
根据本发明可用的组合物可以以各种各样的口服和肠胃外剂量形式给药。对本领域技术人员而言明显的是,以下剂量形式可以包括一种或多种根据本发明可用的组合物作为活性成分。
[0082]
对于由根据本发明可用的组合物制备药物,药学上可接受的载体可以是固体的或液体的。固体形式制剂包括粉剂、片剂、丸剂、胶囊、扁囊剂(cachet)、栓剂和可分散颗粒剂。固体载体可以是还可以用作稀释剂、调味剂、增溶剂、润滑剂、悬浮剂、粘合剂、防腐剂、片剂崩解剂或包膜材料(encapsulating material)的一种或多种物质。
[0083]
在粉剂中,载体是与粉碎的活性组分混合的粉碎固体。在片剂中,活性组分与具有必要的结合能力的载体以适合的比例混合,并压缩成所需形状和大小。适合的载体是碳酸镁、硬脂酸镁、滑石、糖、乳糖、果胶、糊精、淀粉、明胶、黄蓍胶、甲基纤维素、羧甲基纤维素钠、低熔点蜡、可可脂等。术语“制剂”意在包括具有包膜材料作为载体的活性化合物配制品,提供其中活性组分在有或没有载体的情况下被载体包围并因此与其结合的胶囊。类似地,包括扁囊剂和锭剂(lozenge)。片剂、粉剂、胶囊、丸剂、扁囊剂和锭剂可以用作适用于口服给药的固体形式。
[0084]
为了制备栓剂,首先熔化低熔点蜡,诸如脂肪酸甘油酯或可可脂的混合物,并使活性组分均匀地分散在其中,如通过搅拌。然后将熔化的均匀混合物倒入大小适中的模具中,允许其冷却并从而凝固。适用于阴道给药的组合物可以表现为除活性成分之外还含有本领域中已知的适当载体的阴道栓(pessary)、止血塞(tampon)、霜剂、凝胶剂、糊剂、泡沫剂或喷剂。液体制剂包括溶液剂、悬浮剂和乳剂,例如水或水
‑
丙二醇溶液。例如,肠胃外注射液体制剂可以配制为含水聚乙二醇溶液。
[0085]
因此,根据本发明的组合物可以配制用于肠胃外给药(例如,通过注射,例如推注(bolus injection)或持续输注),且可以以单位剂量形式存在于具有添加防腐剂的安瓿、预填装注射器、小体积输注或在多剂量容器中。组合物可以采取下述形式,诸如悬浮剂、溶液剂或者含油或含水载体(vehicle)中的乳剂,且可以含有配制剂(formulation agent),诸如悬浮剂、稳定剂和/或分散剂。可替选地,活性成分可以是通过无菌固体的无菌分离或通过溶液冻干获得的粉末形式,用于在使用前与适合的载体例如无菌无热原水复配(constitution)。
[0086]
可以通过将活性组分溶解在水中并根据需要加入适合的着色剂、调味剂、稳定剂和增稠剂来制备适用于口服使用的含水溶液。可以通过用粘性材料诸如天然或合成胶、树脂、甲基纤维素、羧甲基纤维素钠或其他熟知的悬浮剂将粉碎的活性组分分散在水中来制备适用于口服使用的含水悬浮剂。
[0087]
还包括预期在使用前不久转化为液体形式制剂用于口服给药的固体形式制剂。这样的液体形式包括溶液剂、悬浮剂和乳剂。除活性组分外,这些制剂还可以含有着色剂、调味剂、稳定剂、缓冲剂、人工和天然甜味剂、分散剂、增稠剂、增溶剂等。
[0088]
在本发明的一种实施方式中,局部地或全身地或通过两种途径组合地施用药物。
[0089]
对于给药,在一种实施方式中,可以在含有按重量0.001%至70%的组合物,优选地按重量0.01%至70%的组合物,甚至更优选地按重量0.1%至70%的组合物的配制品中给药本发明的组合物。在一种实施方式中,所给药的组合物的适合量在0.01mg/kg体重至1g/kg体重的范围内。
[0090]
适用于给药的组合物还包括:在调味基质(通常为蔗糖和阿拉伯胶或黄蓍胶)中包括活性剂的锭剂、在惰性基质(如明胶和甘油或蔗糖和阿拉伯胶)中包括活性成分的软锭剂(pastille)以及在适合的液体载体中包括活性成分的漱口剂(mouthwash)。
[0091]
溶液剂或悬浮剂通过常规手段例如用滴管、移液管或喷雾器直接给药至鼻腔。组合物可以提供为单或多剂量形式。在滴管或移液管的后一种情况中,可以由给药适合的预定体积的溶液或悬浮液的患者来实现。在喷雾器的情况下,可以例如通过计量雾化喷雾泵来实现。
[0092]
对呼吸道的给药也可以通过气雾剂的方式实现,其中用合适的推进剂诸如含氯氟烃(cfc)(例如二氯二氟甲烷、三氯氟甲烷或二氯四氟乙烷)、二氧化碳或其它合适的气体在加压包装中提供活性组分。该气雾剂还可以方便地含有表面活性剂,如卵磷脂。药物的剂量可以通过设置计量阀来控制。
[0093]
可替选地,该活性成分可以提供为干粉形式,例如组合物在合适的粉末基质诸如乳糖、淀粉、诸如羟丙基甲基纤维素的淀粉衍生物以及聚乙烯吡咯烷酮(pvp)中的粉末混合物。方便地,粉末载体会在鼻腔内形成凝胶。粉末组合物可以以单位剂量形式存在,例如,如明胶的胶囊或药筒(cartridges),或者是粉末可以通过吸入器从其给药的泡罩包装(blister pack)。
[0094]
在需要时,可以使用适于使活性成分缓释的药物组合物。
[0095]
药物制剂优选为单位剂量形式。在这种形式中,制剂被细分为含有合适量的活性组分的单位剂量。单位剂量形式可以是包装的制剂,该包装含有分立的制剂量,诸如小瓶或安瓿中的包装片剂、胶囊和粉剂。而且,单位剂量形式可以是胶囊、片剂、扁囊剂或锭剂本身,或者其可以是合适数量的这些剂量形式中任意一种的包装形式。用于口服给药的片剂或胶囊和用于静脉内给药和持续输注的液体是优选的组合物。
[0096]
关于配制和给药的技术的其他详细信息可见于最新版的“remington's pharmaceutical sciences(雷明顿药物科学)(maack publishing co.easton,pa.)和remington:the science and practice of pharmacy“,lippincott williams and wilkins。
[0097]
适合配制品和制造它们的方式也在例如lachman等人著写的
“
arzneiformenlehre,paul heinz list,ein lehrbuchf
ü
rpharmazeuten,wissenschaftliche verlagsgesellschaft stuttgart,4.auflage,1985”或”the theory and practice of industrial pharmacy”,varghese publishing house,1987”或“modern pharmaceutics”,james swarbrick编辑,第2版”中公开。
[0098]
以下为具体实施例
[0099]
下文参照以下实施例进一步描述本发明,实施例意在说明而并不限制本发明的范围。除非另有说明,以下实施例中使用的原料和试剂均为市售商品,或者可以通过已知方法制备。实施例中未注明具体条件的实验方法,按照常规条件,例如文献、书本中所述的条件或者生产厂家推荐的方法实现。
[0100]
实施例1纳米黄芩苷的制备
[0101]
1)、向纳米研磨机中,加入黄芩苷50克,水500毫升,助悬剂吐温
‑
20 50毫克,羟丙甲纤维素50毫克,聚乙二醇6000 50毫克,以2000rpm转速研磨20分钟,得到黄芩苷纳米混悬液,黄芩苷粒径分布在50nm~500nm范围内。
[0102]
2)、得到的黄芩苷纳米混悬液在流化床干燥设备干燥,干燥进风温度65℃,干燥至水分含量3%左右,制备得到黄芩苷纳米颗粒。
[0103]
制备得到的黄芩苷纳米颗粒相比未经纳米研磨的黄芩苷,在10分钟20℃的溶解度增加了3倍。
[0104]
实施例2纳米野黄芩苷的制备
[0105]
与实施例1的制备方法基本相同,不同之处在于,将黄芩苷替换为野黄芩苷。野黄芩苷粒径分布在50nm~500nm范围内。
[0106]
制备得到的野黄芩苷纳米颗粒相比未经纳米研磨的野黄芩苷,在10分钟20℃的溶解度增加了3倍
[0107]
实施例3:实施例1得到的黄芩苷纳米颗粒1克(以黄芩苷含量计),羟氯喹10克,混合均匀,得到黄芩苷纳米颗粒羟氯喹组合物,黄芩苷与羟氯喹的比例为1:10。
[0108]
实施例4:实施例1得到的黄芩苷纳米颗粒10克(以黄芩苷含量计),羟氯喹1克,混合均匀,得到黄芩苷纳米颗粒羟氯喹组合物,黄芩苷与羟氯喹的比例为10:1。
[0109]
实施例5:实施例2得到的野黄芩苷纳米颗粒1克(以野黄芩苷含量计),羟氯喹10克,混合均匀,得到野黄芩苷纳米颗粒羟氯喹组合物,野黄芩苷与羟氯喹的比例为1:10。
[0110]
实施例6:实施例2得到的野黄芩苷纳米颗粒10克(以野黄芩苷含量计),羟氯喹1克,混合均匀,得到野黄芩苷纳米颗粒羟氯喹组合物,野黄芩苷与羟氯喹的比例为10:1。
[0111]
实施例7:实施例1得到的黄芩苷纳米颗粒29克(以黄芩苷含量计),羟氯喹21克,混合均匀,得到黄芩苷纳米颗粒羟氯喹组合物,黄芩苷与羟氯喹的比例为29:21。
[0112]
实施例8:实施例2得到的野黄芩苷纳米颗粒29克(以野黄芩苷含量计),羟氯喹21克,混合均匀,得到野黄芩苷纳米颗粒羟氯喹组合物,野黄芩苷与羟氯喹的比例为29:21。
[0113]
实施例9动物在体抗炎活性测试
[0114]
分别设置空白给药组、阴性对照组和不同活性成分给药组。
[0115]
1、试验动物
[0116]
小鼠:c57bl/6j鼠,雄性,体重20g,6
‑
8周龄。所有小鼠均自由取食和饮水,在室温(23
±
2)℃条件下饲养。
[0117]
2、试验方法
[0118]
阴性对照组:未做任何处理的小鼠10只,给予生理盐水
[0119]
建立炎症患病小鼠,并将炎症患病小鼠随机分组,每组10只,给药方案如下:
[0120]
空白给药组:给予生理盐水
[0121]
黄芩苷组:未研磨黄芩苷,按照29mg/kg给药量,灌胃,每日一次,连续给药3日。
[0122]
野黄芩苷组:未研磨野黄芩苷,按照29mg/kg给药量,灌胃,每日一次,连续给药3日。
[0123]
羟氯喹组:羟氯喹,按照21mg/kg给药量,灌胃,每日一次,连续给药3日。
[0124]
组1:未研磨黄芩苷与羟氯喹按10:1比例的混合物,按照310mg/kg给药量,灌胃给药,每日一次,连续给药3日。
[0125]
组2:未研磨野黄芩苷与羟氯喹按10:1比例的混合物,按照310mg/kg给药量,灌胃给药,每日一次,连续给药3日。
[0126]
组3:实施例3得到的黄芩苷纳米颗粒羟氯喹组合物(黄芩苷与羟氯喹比例为1:10),按照24mg/kg给药量,灌胃给药,每日一次,连续给药3日。
[0127]
组4:实施例4得到的黄芩苷纳米颗粒羟氯喹组合物(黄芩苷与羟氯喹比例为10:1),按照310mg/kg给药量,灌胃给药,每日一次,连续给药3日。
[0128]
组5:实施例5得到的野黄芩苷纳米颗粒羟氯喹组合物(野黄芩苷与羟氯喹比例为1:10),按照24mg/kg给药量,灌胃给药,每日一次,连续给药3日。
[0129]
组6:实施例6得到的野黄芩苷纳米颗粒羟氯喹组合物(野黄芩苷与羟氯喹比例为10:1),按照310mg/kg给药量,灌胃给药,每日一次,连续给药3日。
[0130]
组7:实施例7得到的黄芩苷纳米颗粒羟氯喹组合物(黄芩苷与羟氯喹比例为29:21),按照50mg/kg给药量,灌胃给药,每日一次,连续给药3日。
[0131]
组8:实施例8得到的野黄芩苷纳米颗粒羟氯喹组合物(野黄芩苷与羟氯喹比例为29:21),按照50mg/kg给药量,灌胃给药,每日一次,连续给药3日。
[0132]
给药结束后,取血测定炎症细胞因子,测定空白给药组炎症细胞因子约为阴性对照组的3倍,以空白给药组炎症细胞因子均值为100%,计算每组小鼠炎症细胞因子相对均值(即每组小鼠炎症细胞因子值除以空白给药组炎症细胞因子值),结果如下:
[0133]
空白给药组炎症细胞因子均值100%,未研磨黄芩苷组(剂量29mg/kg)炎症细胞因子相对均值92%,未研磨野黄芩苷组(剂量29mg/kg)炎症细胞因子相对均值90%,羟氯喹组(剂量21mg/kg)炎症细胞因子相对均值54%,组1(剂量310mg/kg)炎症细胞因子相对均值55%,组2(剂量310mg/kg)炎症细胞因子相对均值53%,组3(剂量24mg/kg)炎症细胞因子相对均值46%,组4(剂量310mg/kg)炎症细胞因子相对均值47%,组5(剂量24mg/kg)炎症细胞因子相对均值47.5%,组6(剂量310mg/kg)炎症细胞因子相对均值49.2%,组7(剂量50mg/kg)炎症细胞因子相对均值45.8%,组8(剂量50mg/kg)炎症细胞因子相对均值47.5%,相比较空白给药组,未研磨黄芩苷组、未研磨野黄芩苷组仅有轻微的降低炎症因子作用;羟氯喹组、未研磨黄芩苷加羟氯喹组、未研磨野黄芩苷加羟氯喹组有中等强度的降低炎症因子作用,三组之间水平基本一致,未经研磨的黄芩苷或野黄芩苷对羟氯喹的降低炎症因子的作用无明显增强作用;三种比例的黄芩苷纳米粒或野黄芩苷纳米粒加羟氯喹组降低炎症因子的作用更强,超过50%。由此可见,经过纳米研磨的黄芩苷或者野黄芩苷对于羟氯喹的降低
炎症因子作用有明显的促进,而未经研磨的黄芩苷或野黄芩苷没有促进作用。
[0134]
以上所述实施例的各技术特征可以进行任意的组合,为使描述简洁,未对上述实施例中的各个技术特征所有可能的组合都进行描述,然而,只要这些技术特征的组合不存在矛盾,都应当认为是本说明书记载的范围。
[0135]
以上所述实施例仅表达了本发明的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对发明专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。因此,本发明专利的保护范围应以所附权利要求为准。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1