一种构建植物发育分子调控网络的方法及其应用与流程
1.本发明涉及分子生物学技术领域,特别是涉及一种构建植物发育分子调控网络的方法及其应用。
背景技术:
2.mirna是一类长度约为21nt的非编码小分子rna,是真核生物中几乎所有生物过程的关键调控因子。在植物体内,mirna能够通过与靶mrna分子的互补序列结合,使其降解或抑制其翻译,在转录后水平调控靶基因的表达,一直是遗传、发育、干细胞等多个研究领域中的研究热点。例如,mir165/mir166通过影响hd-zip iii转录因子基因的表达,调控sam终止、器官极性改变和维管组织和束间纤维发育缺陷等。同时argonautes(agos)通过竞争性结合mirna165/166调控拟南芥茎端分生组织发育和叶片极性形成。这些结果均表明mirna能够联合上游调控基因和下游靶基因,在植物的胚胎发育、分生组织和器官发生等关键发育过程中发挥重要作用。
3.此外,mirnas通过参与调控植物物质和能量代谢、植物激素信号传导以及逆境响应相关生物学途径,影响植物的发育与形态建成。如microrna165/166能够联合植物激素信号传导途径共同维持拟南芥根部维管束构型的稳定(muraro et al.,2014)。以往研究成果显示几乎所有mirnas均受到多个转录因子的调控,同时mirnas也作用于包括转录因子基因的众多靶基因。不仅如此,转录因子通过特异结合下游基因启动子区的dna顺式作用元件,对基因(包括mirna)的转录起到激活或者抑制等作用,形成“mirna-转录因子-靶基因(mirna)”组成的较为复杂的转录调控网络,参与包括植物生长和发育的多个生物通路。即mirna通过与上游调控基因和下游靶基因构成复杂调控网络,参与植物的物质和能量代谢、激素信号转导和逆境胁迫响应等多生物过程,在转录水平调控植物的生长发育。
4.目前也有利用mirna研究植物生长发育的相关记载,比如,研究报道“integrated analysis of small rna,transcriptome and degradome sequencing provides new insights into floral development and abscission in yellow lupine(lupinus luteus l.)”和“integrated transcriptome,small rna and degradome sequencing approaches provide insights into ascochyta blight resistance in chickpea”中团队均发掘得到了候选的mirna,检测到了差异表达的mirna,虽然进行了降解组检测,找到了部分mirna降解靶基因位点,但是混样后很难确定mirna-靶基因对是否来自相同的生长发育时期,即只是在序列上存在互作关系,存在假阳性。虽然运用qrt-pcr检测到两者表达量存在负相关关系,但是没有对候选mirna-靶基因对在细胞中是否能够互作进行验证,也没有对mirna和靶基因在植物组织中的位置进行明确,存在很高的假阳性概率。利用找到的候选mirna及其靶基因构建了调控网络,但由于关键节点mirna及其靶基因存在很高的假阳性概率,也是不能真正反映植物发育过程,对指导育种的价值不大。
技术实现要素:
5.本发明的目的在于提供一种构建植物发育分子调控网络的方法及其应用,能够精确地实现节点基因和mirna在发育组织中的定位,从而精确地、具有时空特异性地反映植物组织发育过程分子调控的特点。
6.为解决上述技术问题,本发明提供了以下技术方案:
7.本发明提供了一种构建植物发育分子调控网络的方法,所述方法包括以下步骤:
8.对植物组织基因文库进行测序得到mirnas-靶基因对;
9.利用双荧光素酶报告检测技术验证所述mirnas-靶基因对,然后通过荧光原位杂交技术对所述mirnas-靶基因对在植物组织细胞中作用位置进行定位后即可获得所述的植物发育分子调控网络。
10.优选的,所述测序包括转录组测序、小rna测序和降解组测序。
11.优选的,所述转录组测序包括:
12.对植物组织基因文库进行测序、数据分析、筛选得到不同植物组织发育时期差异表达的基因。
13.优选的,所述小rna测序包括:
14.对植物组织基因文库进行测序、数据分析、筛选得到植物相邻发育时期差异表达的mirnas,并预测所述mirnas的靶基因。
15.优选的,所述降解组测序包括:
16.对植物组织基因文库进行测序,将测序得到的cluster tags与rfam数据库进行比对获得未被注释的序列,然后对所述未被注释的序列进行分析获得降解位点,经分析筛选得到所述mirnas-靶基因对。
17.优选的,所述植物组织基因文库为根据植物组织rna构建所得的cdna文库。
18.优选的,所述验证包括如下步骤:
19.利用双荧光素酶检测所述的mirnas-靶基因对,得到检测结果为阳性的mirna-靶基因对。
20.优选的,所述定位包括如下步骤:
21.将所述检测结果为阳性的mirna-靶基因对进行荧光原位杂交,观察mirna与靶基因是否两两都能够定位在样本的同一组织。
22.优选的,根据所述mirna与靶基因的定位位置,运用cytoscape软件绘制即可得到可视化的所述植物发育分子调控网络。
23.本发明还提供了上述的方法或利用该方法构建得到的分子调控网络在指导分子育种中的应用。
24.本发明提供了一种构建植物发育分子调控网络的方法,针对组学鉴定的批量mirna-靶基因对,先后运用双荧光素酶报告检测技术和荧光原位杂交技术验证候选mirna及其靶基因的互作关系并定位其在组织和细胞中的位置,精确地实现了节点基因和关键开关mirna在发育组织中的定位,能够精确地、具有时空特异性地反映植物组织发育过程分子调控的特点,对分子设计育种具有很高的指导意义和应用价值。
附图说明
25.图1为小rna测序和数据分析流程。
26.图2为降解组实验流程图。
27.图3为降解组数据分析流程图。
28.图4为荧光原位杂交fish实验定位ped-mir160a-5p能够在笋芽发育早期的原形成层(箭头1)发挥作用。
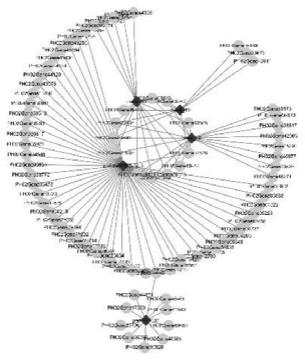
29.图5为毛竹笋芽发育早期和发育中期主要作用因子构成分子调控网络。
30.图6为荧光原位杂交fish实验定位ped-mir166a-3p能够在笋芽发育早期顶端分生组织(箭头1)发挥作用。
31.图7毛竹笋芽萌动期和发育早期主要作用因子构成分子调控网络。
32.图8为荧光原位杂交fish实验定位ped-mir166a-3p能够在笋芽发育中期的维管组织(箭头1)发挥作用。
33.图9毛竹笋芽发育中期和厚竹笋芽发育中期主要作用因子构成分子调控网络。
具体实施方式
34.本发明提供了一种构建植物发育分子调控网络的方法,所述方法包括以下步骤:
35.对植物组织基因文库进行测序得到mirnas-靶基因对;
36.利用双荧光素酶报告检测技术验证所述mirnas-靶基因对,然后通过荧光原位杂交技术对所述mirnas-靶基因对在植物组织细胞中作用位置进行定位后即可获得所述的植物发育分子调控网络。
37.本发明中,所述测序包括转录组测序、小rna测序和降解组测序。所述转录组测序包括:对植物组织基因文库进行测序、数据分析、筛选得到不同植物组织发育时期差异表达的基因。本发明中,所述转录组测序的平台优选为illumina平台。所述数据分析优选的包括如下步骤:将测序所得数据进行过滤得到clean data,与所研究物种参考基因组进行序列比对,得到的mapped data进行插入片段长度检验、随机性检验等文库质量评估;进行可变剪接分析、新基因发掘和基因结构优化等结构水平分析;根据基因在植物组织不同发育时期样品组中的表达量进行差异表达分析。本发明中,所述转录组测序的目的是为了获得植物组织相邻发育时期的差异表达基因。
38.本发明中,所述小rna测序包括:对植物组织基因文库进行测序、数据分析、筛选得到植物相邻发育时期差异表达的mirnas,并预测所述mirnas的靶基因。所述小rna测序的平台优选为illumina平台。所述小rna测序后的数据分析优选的包括如下步骤:小rna测序所得的原始测序数据经质量控制后得到srna,然后对所述的srna进行分类注释获得mirnas,对所述的mirnas进行鉴定分析和表达量分析后筛选差异表达的mirnas。本发明中,所述筛选差异表达的mirnas时优选的根据实际情况选取合适的差异表达分析软件即可。在本发明的具体实施例中,优选的采用deseq2软件进行样品组间的差异表达分析,获得两个生物学条件之间的差异表达mirna集。本发明中,筛选获得差异表达的mirnas后,根据已知mirna和新预测的mirna与对应物种的基因序列信息,用targetfinder软件进行靶基因预测。
39.本发明中,所述降解组测序包括:对植物组织基因文库进行测序,将测序得到的cluster tags与rfam数据库进行比对获得未被注释的序列,然后对所述未被注释的序列进
行分析获得降解位点,经分析筛选得到所述mirnas-靶基因对。所述降解组测序的平台优选为illumina hiseq 2500。所述cluster tags优选为测序得到的原始tags经过去接头和过滤低质量所得。本发明中,所述降解位点的检测优选的采用cleaveland3软件。本发明中,所述降解组测序的目的是为了筛选获得mirnas-靶基因对。
40.本发明中,所述植物组织基因文库为根据植物组织rna构建所得的cdna文库。本发明中所述植物组织优选为植物某一和/或某些组织和细胞。本发明中,所述cdna文库的构建优选的包括如下步骤:(1)用带有oligo(dt)的磁珠富集植物组织的mrna;(2)加入fragmentation buffer将mrna进行随机打断;(3)以mrna为模板,用由碱基“a”、“t”、“c”和“g”组成的六碱基随机引物(random hexamers)合成第一条cdna链,然后加入缓冲液、dntps、rnase h和dna polymerase i合成第二条cdna链,利用ampure xp beads纯化cdna;(4)纯化的双链cdna再进行末端修复、加a尾并连接测序接头,然后用ampure xpbeads进行片段大小选择;(5)最后通过pcr富集得到cdna文库。本发明中,所述提取植物组织rna优选采用试剂盒(购自天根生化科技有限公司)进行提取。
41.本发明中,所述验证包括如下步骤:利用双荧光素酶检测所述的mirnas-靶基因对,得到检测结果为阳性的mirna-靶基因对。本发明中,所述验证的目的是验证mirna是否能够与靶基因结合。本发明中,所述双荧光素酶检测优选的采用双荧光素酶报告基因检测试剂盒。
42.本发明中,所述定位包括如下步骤:将所述检测结果为阳性的mirna-靶基因对进行荧光原位杂交,观察mirna与靶基因是否两两都能够定位在样本的同一组织。本发明对所述荧光原位杂交的方法并没有特殊限定,本发明的实施例中优选采用fish原位杂交试剂盒。
43.本发明根据所述mirna与靶基因的定位位置,运用cytoscape软件绘制即可得到可视化的所述植物发育分子调控网络。本发明中,所述植物发育分子调控网络能够调控植物发育的基因、mirna,还能够解析两者之间的互作关系。
44.本发明还提供了所述的方法或利用该方法构建得到的分子调控网络在指导分子育种中的应用。本发明对所述分子育种并没有特殊限定,优选的包括基因工程育种。
45.本发明中,所用原料、试剂与设备均为已知产品,采用常规市售产品即可。
46.本发明中,所用的实验方法如无特殊限定均为本领域常规的技术手段。
47.为了进一步说明本发明,下面结合实施例对本发明提供的技术方案进行详细地描述,但不能将它们理解为对本发明保护范围的限定。
48.实施例1
49.一、采集植物处于不同发育时期的组织或器官,每个时期样本设置三个重复。
50.二、rna提取:采用天根生化科技(北京)有限公司购买的试剂盒提取上述不同发育时期的组织或器官的rna,具体操作按照试剂盒说明书进行。
51.三、文库构建
52.1.采用agilent bioanalyzer 2100system(agilent technologies,ca,usa)设备检测rna样品的纯度、浓度和完整性。
53.2.样品检测合格后,按如下流程构建文库:
54.(1)用带有oligo(dt)的磁珠富集真核生物mrna;
55.(2)加入fragmentation buffer将mrna进行随机打断;
56.(3)以mrna为模板,用六碱基随机引物(random hexamers)合成第一条cdna链,然后加入缓冲液、dntps、rnase h和dna polymerase i合成第二条cdna链,利用ampure xp beads纯化cdna;
57.(4)纯化的双链cdna再进行末端修复、加a尾并连接测序接头,然后用ampure xp beads进行片段大小选择;
58.(5)最后通过pcr富集得到cdna文库。
59.3.文库构建完成后,使用q-pcr方法对文库的有效浓度(文库有效浓度>2nm)进行准确定量,以保证文库质量。检测合格的文库保存用于后续实验。
60.四、对上述文库进行转录组测序和分析
61.1.将上述检测合格的基因文库利用illumina平台进行测序,得到的数据进行生物信息学分析。
62.2.生物信息学分析
63.2.1生物信息学分析流程概括
64.将下机数据进行过滤得到clean data,与该物种参考基因组进行序列比对,得到的mapped data,进行插入片段长度检验、随机性检验等文库质量评估;进行可变剪接分析、新基因发掘和基因结构优化等结构水平分析;根据基因在不同样品或不同样品组中的表达量进行差异表达分析、差异表达基因功能注释和功能富集等表达水平分析。
65.2.2测序数据及其质量控制
66.基于边合成边测序(sequencing by synthesis,sbs)技术,illumina高通量测序平台对cdna文库进行测序,产出大量的高质量data,称为原始数据(raw data),其大部分碱基质量打分能达到或超过q30。
67.2.2.1测序碱基质量值
68.碱基质量值(quality score或q-score)是碱基识别(base calling)出错的概率的整数映射。通常使用的phred碱基质量值公式为:
69.q=-10
×
log 10pq=-10
×
log 10p
70.其中,p为碱基识别出错的概率。
71.2.2.2测序碱基含量分布
72.碱基类型分布检查用于检测有无at、gc分离现象,由于rna-seq所测的序列为随机打断的cdna片段,因随机性打断及碱基互补配对原则,理论上,g和c、a和t的含量每个测序循环上应分别相等,且整个测序过程稳定不变,呈水平线。
73.2.2.3测序质量控制
74.在进行数据分析之前,首先需要确保这些reads有足够高的质量,以保证后续分析的准确。数据进行严格的质量控制,进行如下过滤方式:(1)去除含有接头的reads;(2)去除低质量的reads(包括去除n的比例大于10%的reads;去除质量值q≤10的碱基数占整条read的50%以上的reads)。
75.经过上述一系列的质量控制之后得到的高质量的clean data,以fastq格式提供。
76.2.3转录组数据与参考基因组序列比对
77.运用hisat2使用基因组作为参考进行序列比对及后续分析。
78.利用stringtie对比对上的reads进行组装和定量。
79.2.4转录组文库质量评估
80.合格的转录组文库是转录组测序的必要条件,为确保文库的质量,从以下3个不同角度对转录组测序文库进行质量评估:(1)通过检验插入片段在基因上的分布,评估mrna片段化的随机性、mrna的降解情况;(2)通过插入片段的长度分布,评估插入片段长度的离散程度;(3)通过绘制饱和度图,评估文库容量和mapped data是否充足。
81.2.5新基因分析
82.2.5.1新基因发掘
83.基于所选参考基因组序列,使用stringtie软件对mapped reads进行拼接,并与原有的基因组注释信息进行比较,寻找原来未被注释的转录区,发掘该物种的新转录本和新基因,从而补充和完善原有的基因组注释信息。过滤掉编码的肽链过短(少于50个氨基酸残基)或只包含单个外显子的序列,发掘得到新基因。
84.2.5.2新基因功能注释
85.使用blast软件将发掘的新基因与nr,swiss-prot,go,cog,kog,pfam,kegg数据库进行序列比对,使用kobas2.0得到新基因的kegg orthology结果,预测完新基因的氨基酸序列之后使用hmmer软件与pfam数据库比对,获得新基因的注释信息。
86.2.6基因表达量分析
87.2.6.1基因表达定量
88.抽取自一个转录本的片段数目与测序数据(或mapped data)量、转录本长度、转录本表达水平都有关,为了让片段数目能真实地反映转录本表达水平,需要对样品中的mapped reads的数目和转录本长度进行归一化。stringtie通过最大流量算法,采用fpkm(fragments per kilobase of transcript per million fragments mapped)作为衡量转录本或基因表达水平的指标,fpkm计算公式如下:
[0089][0090]
其中,cdna fragments表示比对到某一转录本上的片段数目,即双端reads数目;mapped fragments(millions)表示比对到转录本上的片段总数,以10^6为单位;transcript length(kb):转录本长度,以10^3个碱基为单位。
[0091]
2.7差异表达分析
[0092]
基因表达具有时间和空间特异性,在两个不同条件下,表达水平存在显著差异的基因或转录本,称之为差异表达基因(deg)。
[0093]
差异表达分析得到的基因集合叫做差异表达基因集,使用“a_vs_b”的方式命名。根据两(组)样品之间表达水平的相对高低,差异表达基因可以划分为上调基因(up-regulated gene)和下调基因(down-regulated gene)。上调基因在样品(组)b中的表达水平高于样品(组)a中的表达水平;反之为下调基因。上调和下调是相对的,由所给a和b的顺序决定。
[0094]
2.7.1差异表达筛选
[0095]
对于有生物学重复的样本,deseq适用于进行样品组间的差异表达分析,获得两个生物学条件之间的差异表达基因集;对于没有生物学重复的样本,使用ebseq进行差异分
析。
[0096]
在差异表达基因检测过程中,将fold change≥2且fdr《0.01作为筛选标准。差异倍数(fold change)表示两样品(组)间表达量的比值。错误发现率(false discovery rate,fdr)是通过对差异显著性p值(p-value)进行校正得到的。由于转录组测序的差异表达分析是对大量的基因表达值进行独立的统计假设检验,会存在假阳性问题,因此在进行差异表达分析过程中,采用了公认的benjamini-hochberg校正方法对原有假设检验得到的显著性p值(p-value)进行校正,并最终采用fdr作为差异表达基因筛选的关键指标。
[0097]
五、对上述基因文库进行小rna测序和分析
[0098]
实验流程如图1所示,具体如下:
[0099]
1.1利用illumina平台测序得到的原始图像数据文件经碱基识别(base calling)转化为原始测序序列(raw data或raw reads),结果以fastq文件格式存储,其中包含测序序列的序列信息以及其对应的测序质量信息。
[0100]
1.1.1测序碱基质量值
[0101]
碱基质量值(quality score或q-score)是碱基识别(base calling)出错的概率的整数映射。通常使用的phred碱基质量值公式为:
[0102]
q=-10
×
log10pq=-10
×
log10p
[0103]
其中p为碱基识别出错的概率。
[0104]
1.1.2测序数据产出统计
[0105]
测序得到的原始序列含有接头序列或低质量序列,为了保证信息分析的准确性,需要对原始数据进行质量控制,得到高质量序列(即clean reads),原始序列质量控制的标准为:
[0106]
(1)对于每个样本,将质量值低的序列去掉。
[0107]
(2)去除未知碱基n(n为无法识别的碱基)含量大于等于10%的reads;
[0108]
(3)去除没有3’接头序列的reads;
[0109]
(4)剪切掉3’接头序列;
[0110]
(5)去除短于18或长于30个核苷酸的序列;
[0111]
1.2srna分类注释
[0112]
1.2.1ncrna及重复序列注释
[0113]
利用bowtie软件,将clean reads分别与silva数据库、gtrnadb数据库、rfam数据库和repbase数据库进行序列比对,过滤核糖体rna(rrna)、转运rna(trna)、核内小rna(snrna)、核仁小rna(snorna)等ncrna以及重复序列,获得包含mirna的unannotated reads。
[0114]
1.2.2与参考基因组比对
[0115]
利用bowtie软件将unannotated reads与该物种参考基因组进行序列比对,获取在参考基因组上的位置信息,即为mapped reads。
[0116]
1.3mirna分析
[0117]
1.3.1mirna鉴定
[0118]
在已知mirna鉴定方面,将比对到参考基因组的reads与mirbase(v22)数据库中收录的该物种(若暂无收录,则可选其近缘种)mirna的成熟序列及其上游2nt与下游5nt的范
围进行比对,最多允许一个错配,这样鉴定到的reads被认为是鉴定到的已知mirna。
[0119]
mirna转录起始位点多位于基因间隔区、内含子以及编码序列的反向互补序列上,其前体具有标志性的发夹结构,成熟体的形成是由dicer/dcl酶的剪切实现的。针对mirna的生物特征,对于未鉴定到已知mirna的序列,利用mirdeep2软件进行新mirna的预测。
[0120]
利用mirdeep2软件包,通过reads比对到基因组上的位置信息得到可能的前体序列,基于reads在前体序列上的分布信息(基于mirna产生特点,mature,star,loop)及前体结构能量信息(rnafold randfold)采用贝叶斯模型经打分最终实现新mirna的预测。
[0121]
1.3.2mirna碱基偏好性分析
[0122]
dicer酶和dcl酶在识别和切割前体mirna时,5'端首位碱基对u具有很强的偏向性。通过对mirna的碱基偏好性分析,获得典型的mirna碱基比例。
[0123]
1.3.3mirna碱基编辑分析
[0124]
mirna存在转录后碱基的编辑,导致种子序列(seed sequence)改变,进而使得作用的靶基因发生改变。使用isomirid软件检测发生碱基编辑的mirna,isomirid软件首先调用bowtie和前体序列进行第一轮(r0)比对,完全比对的序列作为下轮比对的参考模板。第二轮(r1)比对时,允许一个错配,3'端有一个错配的记为m3,5'端有一个错配的记为m5,中间有一个错配的记为mm。
[0125]
1.3.4mirna家族分析
[0126]
mirna在物种间具有高度保守性,基于序列的相似性对检测到的已知mirna和新mirna进行mirna家族分析,研究mirna在进化中的保守性。
[0127]
1.4mirna表达量分析
[0128]
1.4.1mirna定量
[0129]
对各样本中mirna进行表达量的统计,并用tpm算法[5]对表达量进行归一化处理。tpm归一化处理公式为:
[0130][0131]
公式中,readcount表示比对到某一mirna的reads数目;mapped reads表示比对到所有mirna上的reads数目。
[0132]
1.5mirna差异表达分析
[0133]
1.5.1差异表达mirna筛选
[0134]
利用deseq2检测差异表达mirna,deseq2适用于有生物学重复的实验,可以进行样品组间的差异表达分析,获得两个生物学条件之间的差异表达mirna集。在差异表达mirna检测过程中,使用|log2(fc)|≥1.00;fdr p value≤0.05作为筛选标准。
[0135]
差异表达分析寻找到的基因集合叫做差异表达基因集(deg)。
[0136]
1.5.2差异表达mirna聚类分析
[0137]
对筛选出的差异表达mirna做层次聚类分析,将具有相同或相似表达行为的mirna进行聚类。
[0138]
1.6mirna靶基因预测
[0139]
根据已知mirna和新预测的mirna与对应物种的基因序列信息,用targetfinder软件进行靶基因预测。
[0140]
六、对样本进行降解组测序和分析
[0141]
实验流程如图2,具体步骤如下:
[0142]
1.样品检测和测序
[0143]
(1)通过磁珠捕获植物样本mrna,5’adaptor连接;(2)随机引物和mrna的混合反转录;(3)pcr扩增,完成整个文库制备工作后,构建好的文库用illumina hiseq 2500进行测序。
[0144]
2.信息分析流程
[0145]
原始tags经过去接头和过滤低质量,获得clean tags及cluster tags(clean tags的聚类数据)。将cluster tags和rfam数据库进行比对,注释掉非编码rna,未被注释的序列将用于降解位点分析。降解组分析流程见图3。
[0146]
3.生物信息分析结果
[0147]
3.1基本分析
[0148]
3.1.1测序结果统计
[0149]
原始tags经过去接头和过滤低质量。
[0150]
3.1.2非编码rna注释
[0151]
将cluster tags与基因组和rfam库进行比对,获得与基因组比对的统计结果和非编码rna注释信息,未被注释的序列将用于后续分析。
[0152]
3.2降解位点分析
[0153]
利用mirna库中的已知mirna和小rna项目分析中预测的mirna与对应物种基因转录本序列信息文件,通过cleaveland3软件进行降解位点检测。设置条件p-value《0.05。
[0154]
七、双荧光素酶报告检测
[0155]
1.主要实验试剂如下表1
[0156]
表1主要实验试剂及来源
[0157][0158]
2.实验步骤
[0159]
2.1基因合成及载体构建
[0160]
2.1.1引物设计与合成
[0161]
根据3.2步骤所得mirna-靶基因对中的靶基因序列,运用primer5软件设计引物并合成序列,进行基因扩增。
[0162]
基因扩增
[0163]
1)pcr扩增反应体系如下表2
[0164]
表2pcr扩增反应体系
[0165][0166][0167]
2)pcr扩增反应条件如下表3
[0168]
表3pcr扩增反应条件
[0169][0170]
扩增片段的电泳检测参照质粒提取试剂盒(购自axygen公司,生产批号12123);回收步骤参照dna回收试剂盒(购自天根生化科技有限公司,生产批号dp214-03)说明书。
[0171]
2.1.2载体与目的基因的酶切
[0172]
所用试剂如下表4
[0173]
表4酶切试剂及体系
[0174][0175]
在37℃的条件下酶切1-2h;酶切产物电泳检测;回收步骤参照dna回收试剂盒(购自天根生化科技有限公司,生产批号dp214-03)说明书。
[0176]
2.1.3载体与目的基因的连接
[0177]
连接试剂如下表5
[0178]
表5连接试剂及体系
[0179][0180]
在16℃的条件下反应0.5-1h,保证载体与目的基因的充分连接。
[0181]
2.1.4转化
[0182]
1)将要转化的dna片段加入到装有top10感受态细胞的管中(50μl感受态细胞需要25ngdna),体积应不超过感受态细胞的5%,轻轻旋转几次混匀内容物,冰浴30min。
[0183]
2)将离心管混合物放入加温至42℃的循环水中,热激90s,不要摇动管。
[0184]
3)快速将管转移到冰浴中,使细胞冷却1~2min。
[0185]
4)每管加入200μlsoc液体培养基,用水浴将培养基加温至37℃,然后将管转移到设置为37℃的摇床上,220rpm培养45min,使细胞复苏并表达质粒编码的抗性标记基因。
[0186]
5)将适当体积(每个90mm平板达200μl)已转化的感受态细胞转移到含有相应抗生素的lb培养基上。
[0187]
6)倒置平板,于37℃培养,12~16小时后可出现菌斑。
[0188]
2.1.5菌落pcr验证
[0189]
待平板上长出菌落,随机挑取若干个,进行菌落pcr验证,检测转化子。
[0190]
2.1.6测序验证
[0191]
阳性克隆送本公司测序平台测序验证。
[0192]
2.2质粒转染
[0193]
(1)转染前一天24孔培养板中接种细胞,使转染时细胞密度在70-80%,培养基为
dmem+10%fbs;
[0194]
(2)mirna用depc水溶解至浓度为20μm,然后取mirna(转染细胞的终浓度为50nm)和2μg质粒加至50μl无血清dmem培养基室温孵育5min,另将2μl lipo2000转染试剂加入50μl无血清dmem培养基,将上述两者混合后室温孵育15min,补加无血清dmem培养基至500μl;
[0195]
(3)吸弃皿中培养基,加入上一步制备的转染复合物500μl,37℃培养4-6h;
[0196]
(4)吸弃培养基,加入0.5ml完全培养基,37℃培养48h;
[0197]
(5)转染做5次重复;
[0198]
其中:
[0199]
a.dna的量:lipofectamine 2000=1:1
[0200]
b.细胞密度大约占培养板的80%左右,转染
[0201]
2.3双荧光素酶检测(双萤光素酶报告基因检测试剂盒,rg027)
[0202]
采用双荧光素酶报告基因检测试剂盒操作说明进行此部分操作,此试剂盒中对应试剂。包装清单如下:
[0203]
rg027-1:报告基因细胞裂解液
[0204]
rg027-2:荧光素酶检测试剂
[0205]
rg027-3:海肾荧光素酶检测缓冲液
[0206]
rf027-4:海肾荧光素酶检测底物(100
×
)
[0207]
具体实验方法如下:
[0208]
(1)裂解细胞:24孔板中每孔中加入细胞裂解液(rg027-1)200μl,室温孵育10min,充分裂解细胞;
[0209]
(2)充分裂解后,收裂解液,10000rpm离心5min,取上清作为待测液;
[0210]
(3)融解萤火虫萤光素酶检测试剂(rg027-2)和海肾萤光素酶检测缓冲液(rg027-3),并达到室温。海肾萤光素酶检测底物(100
×
)置于冰浴备用。
[0211]
(4)开启化学发光仪,另取96孔板,取100μl细胞裂解液上清加入96孔发光板中,再加入100μl萤火虫荧光素酶检测工作液(rg027-2),吹吸混匀;
[0212]
(5)上机测发光值,积分时间1s;
[0213]
(6)在完成上述测定萤火虫萤光素酶步骤后,加入海肾荧光素酶检测工作液(rg027-3)100μl,吹吸混匀,上机测发光值,积分时间1s;
[0214]
(7)在以海肾萤光素酶为内参的情况下,用萤火虫萤光素酶测定得到的rlu值除以海肾萤光素酶测定得到的rlu值,根据得到的比值来比较不同样品间目的报告基因的激活程度。
[0215]
八、荧光原位杂交
[0216]
上述检测结果为阳性的mirna-基因对继续进行荧光原位杂交检测,观察候选mirna与基因是否两两都能够定位在样本的同一组织。
[0217]
其中,实验过程中所有的试验仪器见表6
[0218]
表6试验仪器
[0219]
产品名称品牌货号多聚赖氨酸coolabercp8681多聚甲醛阿拉丁p395744
水浴缸leicahi1220正置荧光显微镜leicadm3000fish原位杂交试剂盒歌凡生物c007
[0220]
实验步骤如下:
[0221]
(1)切片、铺片:用眼科镊子镊起植物组织切片轻轻平铺在40~45℃的水面上,借水的张力和水的温度,将略皱的切片自然展平。
[0222]
(2)贴片、烘片:待切片在恒温水面上充分展平后,将蜡片捞到载玻片的中段处倾去载玻片上的余水,置入60~65℃恒温箱内或切片漂烘温控仪的烘箱内烤片15~30分钟,脱去溶化组织间隙的石蜡。
[0223]
(3)石蜡切片脱蜡至水:依次将切片放入二甲苯ⅰ10min-二甲苯ⅱ10min-无水乙醇ⅰ5min-无水乙醇ⅱ5min-95%酒精5min-90%酒精5min-80%酒精5min-70%酒精5min-蒸馏水洗。
[0224]
(4)制作湿盒:用5
×
ssc(ph7.5)(35ml)+甲酰胺(35ml)置于湿盒内。
[0225]
(5)30%h2o21份+9份纯甲醇混合液室温处理10min。depc水洗3次,每次1min;
[0226]
(6)切片置于湿盒内,用0.25%盐酸滴于组织上,常温15min,用depc水洗2次,每次1min(盐酸可中和带电荷的碱性蛋白,降低背景)
[0227]
(7)蛋白酶k覆盖组织,分子杂交仪中37℃20min。蛋白酶k可以消化和暴露被遮蔽的靶核酸,增加探针的结合效率。
[0228]
(8)用0.2%或0.1mol/l甘氨酸洗液洗1min(现用现配),终止蛋白酶k。
[0229]
(9)pbs洗两次,每次1min。
[0230]
(10)用4%pfa多聚甲醛固定组织10min。
[0231]
(11)pbs洗三次,每次1min。
[0232]
(12)用acetic anhydride乙酸酐ph=8.0(乙酰化作用,降低背景)室温洗5min,2次。(乙酸酐溶液配制:每50ml三乙醇胺水溶液中加126ul乙酸酐,现用现配,按比例配制,用多少配多少)。用pbs洗5次,每次1min。
[0233]
(13)用5
×
sscph7.5洗两次,每次1min。
[0234]
(14)切片放置湿盒内,用预杂交液覆盖组织,65℃预杂交1h。
[0235]
(15)100um的探针母液可以1:500-1000稀释,fitc-labeled pube探针覆盖切片,在杂交仪内62-70℃黑暗反应24~72h,反应温度与反应时间视不同的组织与探针而定,建议一般65℃48h。
[0236]
(16)用2
×
ssc ph=7.5,室温洗1次,1min。
[0237]
(17)用甲酰胺加4
×
ssc(ph=4.5)1:1混合液在60℃或65℃洗三次,每次20min。
[0238]
(18)用pbs洗5次,每次1min,室温。
[0239]
(19)用depc处理水1000倍稀释dapi,染核5min。
[0240]
(20)用pbs洗3遍,每遍5min。
[0241]
(21)滴加防淬灭剂,盖盖玻片,指甲油封片,荧光显微镜观察;探针的位置即为目的基因或者mirna的作用位置。
[0242]
上述检测阳性的mirna-基因对存在一个mirna对应多个靶基因或者多个mirna对应一个靶基因的情况,因而组成复杂的调控网络,运用cytoscape软件绘制可视化的调控网
络。
[0243]
实施例2
[0244]
利用实施例1所述的构建植物分子调控网络的方法构建毛竹发育早期原形成层的分子调控网络。
[0245]
其中,毛竹发育早期笋芽样本中鉴定到137个mirnas,包括120个已知和17个新mirnas;基因38652个。而发育中期笋芽样本鉴定到130个mirna,包括115个已知和15个新mirnas;基因39584个。表达量分析鉴定到在两组样本中差异表达mirna30个,差异表达基因61个;降解组测序分析鉴定到101组mirna-靶基因对。双荧光素酶报告检测验证65组mirna-靶基因对能够在细胞内进行互作。荧光原位杂交fish实验定位ped-mir160a-5p等能够在笋芽发育中期的原形成层发挥作用,结果如图4,构建毛竹发育早期原形成层发育分子调控网络1个,如图5。
[0246]
实施例3
[0247]
利用实施例1所述的构建植物分子调控网络的方法构建毛竹发育早期笋芽顶端分生组织的分子调控网络。
[0248]
其中,在毛竹萌动期样本中鉴定到129个mirnas,包括117个已知和12个新mirnas;基因38462个。而发育早期样本鉴定到137个mirna,包括120个已知和17个新mirnas;基因38652个。表达量分析鉴定到在两组样本中差异表达mirna 26个,差异表达基因75个;降解组测序分析鉴定到98组mirna-靶基因对。双荧光素酶报告检测验证56组mirna-靶基因对能够在细胞内进行互作。荧光原位杂交fish实验定位ped-mir166a-3p等能够在笋芽发育早期该时期的顶端分生组织发挥作用,结果如图6,构建毛竹笋芽发育早期顶端分生组织的分子调控网络1个,如图7。
[0249]
实施例4
[0250]
利用实施例1所述的构建植物分子调控网络的方法构建毛竹发育中期维管的分子调控网络。
[0251]
其中,采集处于发育中期的毛竹和厚竹笋芽,通过小rna测序、转录组测序和分析,在毛竹发育中期笋芽样本中鉴定到130个mirna,包括115个已知和15个新mirnas;基因39584个。在厚竹发育中期笋芽样本中鉴定到146个mirnas,包括110已知和36新mirnas,基因38627个。降解组测序分析鉴定到86组mirna-靶基因对。双荧光素酶报告检测验证67组mirna-靶基因对能够在细胞内进行互作。荧光原位杂交fish实验定位ped-mir166a-3p等能够在毛竹和厚竹笋芽该时期的维管组织发挥作用,结果如图8,构建毛竹笋芽发育中期维管的分子调控网络1个,如图9。
[0252]
以上所述仅为本发明的实施例,并非因此限制本发明的专利范围,凡是利用本发明说明书内容所作的等效结构或等效流程变换,或直接或间接运用在其他相关的技术领域,均同理包括在本发明的专利保护范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1