一种预测与新型冠状病毒蛋白质相互作用的药物的方法
1.本发明属于基于半监督机器学习进行分类任务的技术领域,尤其涉及一种预测与新型冠状病毒(sars-cov-2)蛋白质相互作用的药物的方法。
背景技术:
2.由新型冠状病毒(sars-cov-2)引起的新型冠状病毒肺炎(covid-19)在全世界内的持续大流行正在威胁着人类健康。目前,抵抗covid-19的最好方法是接种疫苗,用于阻止sars-cov-2的传播。到目前为止,全世界已有数千项已注册的抗covid-19 治疗的临床试验。许多研究工作集中于研究和验证sars-cov-2蛋白质作为抗病毒药物的靶点。尽管做出了大量的努力,但是目前还没有经过验证的可用于治疗covid-19的抗病毒药物,也没有批准任何治疗covid-19的药物。 sars-cov-2的不断变种使得研究人员难以制定全面的治疗策略,并且正在进行的临床试验也远远地偏离现实。因此,需要一种能够立即应用于患者的快速药物应用策略。目前,解决这一问题的方法之一是在所谓的“药物再利用”,重新利用已有的商用药物分子来治疗不同的病症。许多研究人员正在研究治疗 sars-cov-2感染的不同策略,并正在探索药物的再利用。许多临床试验已经完成,但仍然没有发现任何重新评估的药物可以有效的治疗covid-19。优先考虑已批准的药物对于针对covid-19的快速的临床试验至关重要。
3.尽管在过去二十年中,对生物医学和药物研发的投资大幅增加,但美国食品和药物管理局(fda)批准的年度新疗法数量仍然相对稳定且有限。最近的一项研究估计,制药公司在2015年花费了26亿美元,高于2003年的8.02亿美元,用于开发fda批准的新型化学实体药物。药物再利用作为一种有效的药物发现技术,是一种应对眼前全球挑战的新兴策略。与从头药物发现和随机临床试验相比,药物再利用作为现有药物的有效药物发现策略,可以显着缩短时间并降低成本。然而,药物再利用的实验方法既昂贵又耗时。随着研究人员对生物分子信息的存储技术日益成熟,越来越多的生物分子以数据的形式存储在数据库中。这为基于计算方法的药物再利用提供了数据基础。并且,由于机器学习在数据分类和回归中优异的性能表现,越来越多的研究人员利用机器学习进行药物-靶点相互作(dti)预测,实现基于计算方法的药物再利用。
4.在gansdta:predicting drug-target binding affinity using gans(zhao l, wang j,pang l,et al.gansdta:predicting drug-target binding affinity usinggans[j].frontiers in genetics,2020,10:1243-.)论文中提出了使用对抗神经网络单独进行药物simle特征的提取和蛋白质特征的提取。然而,该方法只单独的对药物和蛋白质特征进行提取,而没有考虑分类器或回归模型实际最终使用到的药物-蛋白质对的整体特征提取。
技术实现要素:
[0005]
本发明的目的是提供一种预测与新型冠状病毒蛋白质相互作用的药物的方法,所
述方法使用随机欠采样解决正负样本不平衡问题;使用自监督学习进行药物-蛋白质对特征的提取,解决样本信息丢失问题。所述方法基于冠状病毒蛋白质预测与sars-cov-2蛋白质相互作用的药物。使用随机森林作为分类器模型进行药物-蛋白质对分类预测。
[0006]
为了从药物-蛋白质对特征中学习到整体的特征信息,本发明提出了使用自监督学习的方法从没有标签的药物-蛋白质对中学习特征信息,从而有效地学习到药物-蛋白质对的整体特征。
[0007]
本发明的目的至少通过如下技术方案之一实现。
[0008]
一种预测与新型冠状病毒蛋白质相互作用的药物的方法,包括如下步骤:
[0009]
s1、根据存在已知相互作用关系的药物和蛋白质,分别获取药物的简化分子线性输入规范(simplified molecular input line entry system,smiles)和已知蛋白质的氨基酸序列信息;获取冠状病毒蛋白质的氨基酸序列信息;计算药物的pubchem分子指纹和蛋白质的氨基酸突变信息特征向量;
[0010]
s2、将药物和蛋白质两两组合为药物-蛋白质对,得到药物-已知蛋白质对和药物-冠状病毒蛋白质对,并根据药物的pubchem分子指纹和蛋白质的氨基酸突变信息特征向量拼接成药物-已知蛋白质对特征以及药物-冠状病毒蛋白质对特征,由药物-已知蛋白质对特征组成未平衡数据集并进行正负样本平衡,生成平衡数据集;
[0011]
s3、使用未平衡数据集训练自监督学习特征提取器,得到已训练的特征提取器;
[0012]
s4、将平衡数据集和由药物-冠状病毒蛋白质对特征组成的冠状病毒数据集输入已训练的特征提取器中,分别生成分类器数据集和待预测冠状病毒数据集;
[0013]
s5、使用分类器数据集训练分类器,得到已训练的分类器;
[0014]
s6、将待预测数据集输入到已训练的分类器中获取预测结果。
[0015]
进一步地,步骤s1中,从数据库中获取已知的药物-蛋白质相互作用对、已知的药物-蛋白质相互作用对中药物的smiles和已知蛋白质的氨基酸序列;排除不存在的药物-蛋白质相互作用关系的药物或已知蛋白质,确保已知蛋白质至少与一种药物存在相互作用关系、药物至少与一种已知蛋白质存在相互作用关系;已知蛋白质从功能上被分成了四种不同功能的蛋白质:酶、离子通道、 g蛋白偶联受体及核受体;为了提高本发明的泛化能力,将四种不同功能的已知蛋白质与药物构成的样本构成药物-已知蛋白质数据集;
[0016]
获取冠状病毒的物种信息,根据不同物种的冠状病毒获取对应的冠状病毒蛋白质的氨基酸序列信息。
[0017]
进一步地,根据药物-已知蛋白质数据集中的药物数据,使用药物的 smiles计算药物的pubchem分子指纹;
[0018]
药物分子的pubchem分子指纹描述药物中是否存在某个分子结构,如果存在某个分子结构,则对应的特征位编码为1,否则对应的特征位编码为0;通过使用生物信息计算软件包,利用药物的smiles计算得到881维的 pubchem分子指纹。
[0019]
进一步地,特异性得分矩阵中存储着在氨基酸序列某个位置的氨基酸突变为其他氨基酸的概率;
[0020]
通过使用美国国家生物信息中心(national center for biotechnologyinformation search database,ncbi)开发的blast工具,使用和药物存在已知相互作用关系的已知蛋白质和冠状病毒蛋白质的氨基酸序列分别计算和药物存在已知相互作用关系
的已知蛋白质和冠状病毒蛋白质的特异性得分矩阵,并通过计算k间隔二元组得分(saini h,raicar g,lal s p,et al.protein foldrecognition using genetic algorithm optimized voting scheme and profilebigram[j].journal of software,2016,11(8):756-767.)对特异性得分矩阵进行编码,得到400维的蛋白质的氨基酸突变信息特征向量,提取特异性得分矩阵中和药物存在已知相互作用关系的已知蛋白质和冠状病毒蛋白质的氨基酸突变信息,1《=k《20。
[0021]
进一步地,步骤s2中,将药物和已知蛋白质两两组合为药物-蛋白质对,得到药物-已知蛋白质对和药物-冠状病毒蛋白质对,并根据药物pubchem分子指纹和蛋白质的氨基酸突变信息特征向量拼接成药物-已知蛋白质对特征以及药物-冠状病毒蛋白质对特征,其中将已知相互作用关系的药物-已知蛋白质对特征为正样本,将未知相互作用关系的药物-已知蛋白质对特征为负样本;混合正负样本并打乱样本顺序得到未平衡数据集,生成的未平衡数据集中的正负样本数量极度不平衡,负样本数量远远大于正样本数量;
[0022]
使用随机欠采样方法从负样本中随机选择与正样本相同数量的负样本,从而解决未平衡数据集中样本不平衡问题。
[0023]
进一步地,步骤s3中,自监督学习特征提取器模型包括四个卷积模块和一个全连接模块;每个卷积模块中包括多个基础卷积模块,基础卷积模块包括一个卷积层、batchnorm层和relu激活函数层;
[0024]
其中,第一卷积模块包括第一基础卷积模块、第二基础卷积模块、第三基础卷积模块以及最大池化层;第二卷积模块包括第四基础卷积模块、第五基础卷积模块、第六基础卷积模块以及第一平均池化层;第三卷积模块包括第七基础卷积模块、第八基础卷积模块、第九基础卷积模块以及第二平均池化层;第四卷积模块包括第十基础卷积模块、第十一基础卷积模块、第十二基础卷积模块;全连接模块包括一个全局平均池化层和一个线性全连接层。
[0025]
进一步地,步骤s3的步骤如下:
[0026]
s3.1、将未平衡数据集的样本特征进行处理,保留原始样本特征赋予标签 0,样本特征向量中的特征值向左循环移动1/3赋予标签1,样本特征向量中的特征值向右循环移动1/3赋予标签2,翻转样本特征向量中的特征值赋予标签 3,从而得到新的带标签的用于训练自监督特征提取器的数据集;将训练自监督特征提取器的数据集拆分为8:2比例的特征提取器训练集和特征提取器测试集;
[0027]
s3.2、使用步骤s3.1中得到的特征提取器训练集训练特征提取器,使用预测值与标签之间交叉熵作为提取器训练的损失,并使用随机梯度下降作为优化函数,训练提取器直至交叉熵收敛至极小值,并选择在特征提取器测试集上准确率最高的特征提取器作为已训练的特征提取器。
[0028]
进一步地,步骤s4中,分别将平衡数据集和冠状病毒数据集输入到已训练的特征提取器中,通过已训练的特征提取器中的前两个卷积模块将原有样本特征转换并输出新学习到的样本特征,分别得到用于训练分类器的分类器数据集和待预测冠状病毒数据集,将分类器数据集按8:2的比例随机拆分为分类器训练集和分类器测试集。
[0029]
进一步地,步骤s5中,选用随机森林模型作为分类器,通过重复对分类器训练集有放回地抽取训练集大小相同的子数据集训练决策树,直至决策树数量达到训练要求的数量
为止,得到构造好的随机森林分类器,将构造好的随机森林分类器在分类器测试集上进行评估,根据评估结果的准确率对分类器的性能进行评估。
[0030]
进一步地,步骤s6中,将药物-冠状病毒蛋白质对新特征作为预测目标,预测与所有物种的冠状病毒蛋白质存在已知相互作用关系的潜在药物,从而预测可能与新型冠状病毒(sars-cov-2)蛋白质存在相互作用关系的药物。
[0031]
相比与现有技术,本发明的优点在于:
[0032]
本发明提出了一种基于半监督学习预测与sars-cov-2蛋白质相互作用的药物的可行方法,通过使用随机欠采样方法处理正负样本不平衡的问题,使用自监督学习方法进行药物-蛋白质对特征的提取,解决样本信息丢失问题。使用随机森林进行药物-蛋白质对分类预测,根据预测结果获得潜在的药物。本发明可以在使用随机欠采样的情况下,仍然能够通过自监督学习提取样本特征,弥补了随机欠采样导致的样本丢失带来的问题,具有更高的模型可信度和预测准确率。
附图说明
[0033]
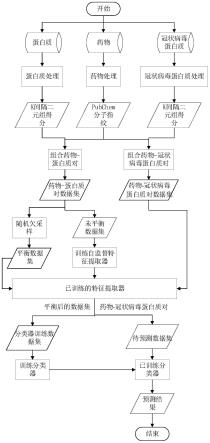
图1为本发明实施例中一种预测与新型冠状病毒蛋白质相互作用的药物的方法的流程示意图。
具体实施方式
[0034]
为使本发明的目的、技术方案和优点更加清楚,下面结合附图对本发明的具体实施方式进行详细说明。这些优选实施方式的示例在附图中进行了例示。附图中所示和根据附图描述的本发明的实施方式仅仅是示例性的,并且本发明并不限于这些实施方式。
[0035]
在此,还需要说明的是,为了避免因不必要的细节而模糊了本发明,在附图中仅仅示出了与根据本发明的方案密切相关的结构和/或处理步骤,而省略了关系不大的其他细节。
[0036]
实施例1:
[0037]
一种预测与新型冠状病毒蛋白质相互作用的药物的方法,如图1所示,包括如下步骤:
[0038]
s1、根据存在已知相互作用关系的药物和蛋白质,分别获取药物的简化分子线性输入规范(simplified molecular input line entry system,smiles)和已知蛋白质的氨基酸序列信息;获取冠状病毒蛋白质的氨基酸序列信息;计算药物的pubchem分子指纹和蛋白质的氨基酸突变信息特征向量;
[0039]
从数据库中获取已知的药物-蛋白质相互作用对、已知的药物-蛋白质相互作用对中药物的smiles和已知蛋白质的氨基酸序列;排除不存在的药物-蛋白质相互作用关系的药物或已知蛋白质,确保已知蛋白质至少与一种药物存在相互作用关系、药物至少与一种已知蛋白质存在相互作用关系;已知蛋白质从功能上被分成了四种不同功能的蛋白质:酶、离子通道、g蛋白偶联受体及核受体;为了提高本发明的泛化能力,将四种不同功能的已知蛋白质与药物构成的样本构成药物-已知蛋白质数据集;
[0040]
获取冠状病毒的物种信息,根据不同物种的冠状病毒获取对应的冠状病毒蛋白质的氨基酸序列信息。
[0041]
根据药物-已知蛋白质数据集中的药物数据,使用药物的smiles计算药物的pubchem分子指纹;
[0042]
药物分子的pubchem分子指纹描述药物中是否存在某个分子结构,如果存在某个分子结构,则对应的特征位编码为1,否则对应的特征位编码为0;通过使用生物信息计算软件包,利用药物的smiles计算得到881维的 pubchem分子指纹。
[0043]
本实施例中,通过使用pybiomed(dong,yao,zj,et al.pybiomed:a pythonlibrary for various molecular representations of chemicals,proteins and dnas andtheir interactions[j].j cheminformatics,2018.)生物信息计算软件包利用药物的smlies计算得到pubchem分子指纹。
[0044]
特异性得分矩阵中存储着在氨基酸序列某个位置的氨基酸突变为其他氨基酸的概率;
[0045]
通过使用美国国家生物信息中心(national center for biotechnologyinformation search database,ncbi)开发的blast工具,使用和药物存在已知相互作用关系的已知蛋白质和冠状病毒蛋白质的氨基酸序列分别计算和药物存在已知相互作用关系的已知蛋白质和冠状病毒蛋白质的特异性得分矩阵,并通过计算k间隔二元组得分(saini h,raicar g,lal s p,et al.protein foldrecognition using genetic algorithm optimized voting scheme and profilebigram[j].journal of software,2016,11(8):756-767.)对特异性得分矩阵进行编码,得到400维的蛋白质的氨基酸突变信息特征向量,提取特异性得分矩阵中和药物存在已知相互作用关系的已知蛋白质和冠状病毒蛋白质的氨基酸突变信息,1《=k《20。
[0046]
本实施例中,将蛋白质氨基酸序列信息上传至possum(wang j,yang b etal.possum:a bioinformatics toolkit for generating numerical sequence featuredescriptors based on pssm profiles.bioinformatics 2017;33(17):2756-2758.)服务器中计算氨基酸序列的特异性得分矩阵和k间隔二元组得分(k取值为1),得到蛋白质氨基酸序列的进化信息特征。
[0047]
s2、将药物和蛋白质两两组合为药物-蛋白质对,得到药物-已知蛋白质对和药物-冠状病毒蛋白质对,并根据药物的pubchem分子指纹和蛋白质的氨基酸突变信息特征向量拼接成完整1281维的药物-已知蛋白质对特征以及药物
‑ꢀ
冠状病毒蛋白质对特征,由药物-已知蛋白质对特征组成未平衡数据集并进行正负样本平衡,生成平衡数据集;
[0048]
将药物和已知蛋白质两两组合为药物-蛋白质对,得到药物-已知蛋白质对和药物-冠状病毒蛋白质对,并根据药物pubchem分子指纹和蛋白质的氨基酸突变信息特征向量拼接成药物-已知蛋白质对特征以及药物-冠状病毒蛋白质对特征,其中将已知相互作用关系的药物-已知蛋白质对特征为正样本,将未知相互作用关系的药物-已知蛋白质对特征为负样本;混合正负样本并打乱样本顺序得到未平衡数据集,生成的未平衡数据集中的正负样本数量极度不平衡,负样本数量远远大于正样本数量;
[0049]
使用随机欠采样方法从负样本中随机选择与正样本相同数量的负样本,从而解决未平衡数据集中样本不平衡问题。
[0050]
s3、使用未平衡数据集训练自监督学习特征提取器,得到已训练的特征提取器;
[0051]
自监督学习特征提取器模型包括四个卷积模块和一个全连接模块;每个卷积模块
中包括多个基础卷积模块,基础卷积模块包括一个卷积层、batchnorm 层和relu激活函数层;
[0052]
其中,第一卷积模块1包括第一基础卷积模块101、第二基础卷积模块102、第三基础卷积模块103以及最大池化层;第二卷积模块2包括第四基础卷积模块204、第五基础卷积模块205、第六基础卷积模块206以及第一平均池化层 21;第三卷积模块3包括第七基础卷积模块307、第八基础卷积模块308、第九基础卷积模块309以及第二平均池化层32;第四卷积模块4包括第十基础卷积模块410、第十一基础卷积模块411、第十二基础卷积模块412;全连接模块包括一个全局平均池化层和一个线性全连接层。
[0053]
本实施例中,第一基础卷积模块101中的卷积层参数设定如下:输入通道数为1,输出通道数为192,卷积核大小为1*5。第二基础卷积模块102中的卷积层参数设置包括:输入通道数为192,输出通道数为160,卷积核大小为1*1。第三基础卷积模块103中的卷积层参数设置包括:输入通道数为160,输出通道数为96,卷积核大小为1*1的卷积层。最大池化层的参数设定如下:内核大小为1*3,步长为2。第四基础卷积模块204的卷积层参数设定如下:输入通道数为96,输出通道数为192,卷积内核大小为1*5。第五基础卷积模块205 的卷积层参数设定如下:输入通道数为192,输出通道数为192,卷积核大小为1*1。第六基础卷积模块206的卷积层参数设定如下:输入通道数为192,输出通道数为192,卷积内核大小为1*1。第一平均池化层21的参数设定如下:内核大小为1*3,步长为2。第七基础卷积模块307中的卷积层参数设定如下:输入通道数为192,输出通道数为192,卷积内核大小为1*3。第八基础卷积模块308中的卷积层参数设定如下:输入通道数为192,输出通道数为192,卷积内核大小为1*1。第九基础卷积模块309中的卷积层参数设定如下:输入通道数为192,输入通道数为192,卷积内核大小为1*1。第二平均池化层32的参数设定如下:内核大小为1*3,步长为2。第十基础卷积模块410的参数设定如下:输入通道数为192,输出通道数为192,卷积内核大小为1*3。第十一基础卷积模块411的参数设定如下:输入通道数为192,输出通道数为192,卷积内核大小为1*1。第十二基础卷积模块412的参数设定如下:输入通道为 192,输出通道数位192,卷积核大小为1*1。
[0054]
步骤s3的步骤如下:
[0055]
s3.1、将未平衡数据集的样本特征进行处理,保留原始样本特征赋予标签 0,样本特征向量中的特征值向左循环移动1/3赋予标签1,样本特征向量中的特征值向右循环移动1/3赋予标签2,翻转样本特征向量中的特征值赋予标签 3,从而得到新的带标签的用于训练自监督特征提取器的数据集;将训练自监督特征提取器的数据集拆分为8:2比例的特征提取器训练集和特征提取器测试集;
[0056]
s3.2、使用步骤s3.1中得到的特征提取器训练集训练特征提取器,使用预测值与标签之间交叉熵作为提取器训练的损失,并使用随机梯度下降作为优化函数,训练提取器直至交叉熵收敛至极小值,并选择在特征提取器测试集上准确率最高的特征提取器作为已训练的特征提取器。
[0057]
s4、将平衡数据集和由药物-冠状病毒蛋白质对特征组成的冠状病毒数据集输入已训练的特征提取器中,分别生成分类器数据集和待预测冠状病毒数据集;
[0058]
分别将平衡数据集和冠状病毒数据集输入到已训练的特征提取器中,通过已训练的特征提取器中的前两个卷积模块将原有样本特征转换并输出新学习到的样本特征,分别
得到用于训练分类器的分类器数据集和待预测冠状病毒数据集,将分类器数据集按8:2的比例随机拆分为分类器训练集和分类器测试集。
[0059]
本实施例中,原有样本特征通过卷第一卷积模块(1)和第二卷积模块(2) 处理后,其特征结构为[192,m],其中m为单通道特征长度,192为通道数量。为了能够在随机森林模型上使用该提取后的特征,求所有通道特征的平均值作为最终的特征,其特征结构为[1,m]。
[0060]
s5、使用分类器数据集训练分类器,得到已训练的分类器;
[0061]
选用随机森林模型作为分类器,通过重复对分类器训练集有放回地抽取训练集大小相同的子数据集训练决策树,直至决策树数量达到训练要求的数量为止,得到构造好的随机森林分类器,将构造好的随机森林分类器在分类器测试集上进行评估,根据评估结果的准确率对分类器的性能进行评估。
[0062]
s6、将待预测数据集输入到已训练的分类器中获取预测结果;
[0063]
将药物-冠状病毒蛋白质对新特征作为预测目标,预测与所有物种的冠状病毒蛋白质存在已知相互作用关系的潜在药物,从而预测可能与新型冠状病毒 (sars-cov-2)蛋白质存在相互作用关系的药物。
[0064]
验证与分析
[0065]
本实施例中,为了评价本发明中提出的平衡样本数量差的方法在预测药物
ꢀ‑
蛋白质相互作用的效果,将其用于真实数据集进行实验,实验使用的数据集为yamanishi等人提出的enzyme数据集(yamanishi y,araki m,gutteridge a, honda w,kanehisa m.prediction of drug-target interaction networks from theintegration of chemical and genomic spaces.bioinformatics.2008jul 1;24(13):i232-40.)。该数据集中存在2903个可获得特征的正样本,存在288127 个可获得特征的负样本。分类器选用随机森林最为分类器,并使用scikit-learn 机器学习python包中默认的模型参数配置。本实施例中,实验采用五重交叉验证进行模型方法的评估。
[0066]
本发明方法与纯粹的随机欠采样方法的对比结果如下表1所示。
[0067]
表1
[0068] 随机欠采样自监督学习+随机欠采样auc值0.942660.95361准确率0.872200.89889f1值0.868630.89529
[0069]
通过比较可知,与纯粹的随机欠采样方法相比,本发明方法的结果在roc 曲线线下面积、预测结果准确率及f1值都远高于随机欠采样。相较于纯粹的随机欠采样方法,本发明可以得到更好的结果,证明本发明是有效果的。
[0070]
表2为当前预测结果得分最高的前10对药物-冠状病毒蛋白质对。
[0071]
表2
[0072]
marcu a,grant jr,sajed t,johnson d,li c,sayeeda z,assempour n,iynkkarani,liu y,maciejewski a,gale n,wilson a,chin l,cummings r,le d,pon a, knox c,wilson m.drugbank 5.0:a major update to the drugbank database for2018.nucleic acids res.2018jan 4;46(d1):d1074-d1082.doi: 10.1093/nar/gkx1037.)数据库中获取的数据,其中,已知相互作用关系的药物
‑ꢀ
蛋白质对有28178对,并作为正样本集。随机生成的815340个药物-蛋白质对负样本组成负样本集,并通过组合正样本集和负样本集得到本实施例中的实验数据集。
[0083]
本发明方法与纯粹的随机欠采样方法的对比结果如下表3所示。
[0084]
表3
[0085] 随机欠采样自监督学习+随机欠采样auc值0.999690.99956准确率0.998500.99864f1值0.998450.99860
[0086]
通过比较可知,与纯粹的随机欠采样方法相比,本发明方法的结果在预测结果准确率及f1值都远高于随机欠采样。相较于纯粹的随机欠采样方法,本发明可以得到更好的结果,证明本发明是有效果的。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1