细胞数据注释方法、装置、设备及介质与流程
1.本发明一般涉及生物信息分析技术领域,具体涉及一种细胞数据注释方法、装置、设备及介质。
背景技术:
2.随着生物信息技术的不断发展,空间转录组技术已经广泛应用到各类医学研究中,例如肿瘤研究、神经科学、发育生物学、分子病理学等不同领域。在医学研究过程中,为了基于空间转录组细胞数据进行基因差异表达分析、细胞发育轨迹分析、基因本体(gene ontology,go)富集分析等研究,需要对空间转录组细胞数据进行细胞注释。
3.目前,相关技术中可以采用分类训练的方法基于参考数据集构建模型,然后通过采用构建的模型预测待注释数据集的细胞类型。
4.然而该方法在判断细胞类型的过程中所利用的参考数据集存在特征片面的问题,这也就导致对细胞类型注释的准确度较低。
技术实现要素:
5.鉴于现有技术中的上述缺陷或不足,期望提供一种细胞数据注释方法、装置、设备及介质,能够基于特征较为全面的细胞数据获得待预测转录组的细胞注释结果,从而提高了空间转录组的细胞注释结果的准确性。所述技术方案如下:
6.根据本技术的一个方面,提供了一种细胞数据注释方法,该方法包括:
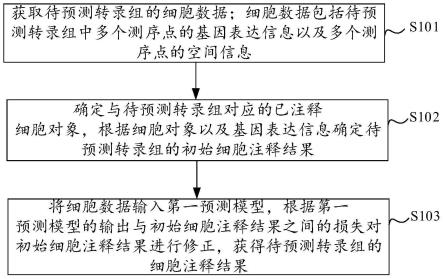
7.获取待预测转录组的细胞数据;所述细胞数据包括所述待预测转录组中多个测序点的基因表达信息以及所述多个测序点的空间信息;
8.确定与所述待预测转录组对应的已注释细胞对象,根据所述细胞对象以及所述基因表达信息确定所述待预测转录组的初始细胞注释结果;
9.将所述细胞数据输入第一预测模型,根据所述第一预测模型的输出与所述初始细胞注释结果之间的损失对所述初始细胞注释结果进行修正,获得所述待预测转录组的细胞注释结果。
10.根据本技术的另一方面,提供了一种细胞数据注释装置,该装置包括:
11.获取模块,用于获取待预测转录组的细胞数据;所述细胞数据包括所述待预测转录组中多个测序点的基因表达信息以及所述多个测序点的空间信息;
12.处理模块,用于确定与所述待预测转录组对应的已注释细胞对象,根据所述细胞对象以及所述基因表达信息确定所述待预测转录组的初始细胞注释结果;
13.细胞注释模块,用于将所述细胞数据输入第一预测模型,根据所述第一预测模型的输出与所述初始细胞注释结果之间的损失对所述初始细胞注释结果进行修正,获得所述待预测转录组的细胞注释结果。
14.根据本技术的另一方面,提供了一种计算机设备,包括存储器、处理器以及存储在存储器上并可在处理器上运行的计算机程序,该处理器执行该程序时实现如上述的细胞数
据注释方法。
15.根据本技术的另一方面,提供了一种计算机可读存储介质,其上存储有计算机程序,该计算机程序用于实现如上述的细胞数据注释方法。
16.根据本技术的另一方面,提供了一种计算机程序产品,其上包括指令,该指令被执行时实现如上述的细胞数据注释方法。
17.本技术实施例中提供的细胞数据注释方法、装置、设备及介质,通过确定与待预测转录组对应的已注释细胞对象,并根据细胞对象以及基因表达信息确定待预测转录组的初始细胞注释结果,在无需任何人工参与注释的情况下,能够获取到待预测转录组的细胞注释结果的指导信息,并且通过第一预测模型对细胞数据进行预测,根据第一预测模型的输出与初始细胞注释结果之间的损失对初始细胞注释结果进行修正,有效地融合了多个测序点的基因表达信息以及多个测序点的空间信息,能够结合指导信息进行自监督修正。一方面,空间转录组的空间信息能够表征空间转录组中各个测序点的空间分布特征,基因表达信息能够表征各个测序点的基因特征。基于空间信息以及基因表达信息确定空间转录组的细胞注释结果,相比于现有技术而言,结合了更为全面的特征来确定细胞注释结果,也使得本技术所提供方法所确定的细胞注释结果的准确度相比于现有技术有明显提升。另一方面,根据第一预测模型的预测损失对细胞注释结果进行进一步的修正,借助第一预测模型的自监督学习算法,实现了细胞注释结果准确度的进一步提升。
18.本发明附加的方面和优点将在下面的描述中部分给出,部分将从下面的描述中变得明显,或通过本发明的实践了解到。
附图说明
19.通过阅读参照以下附图所作的对非限制性实施例所作的详细描述,本技术的其它特征、目的和优点将会变得更明显:
20.图1为本技术实施例提供的细胞数据注释的应用系统的系统架构图;
21.图2为本技术实施例提供的细胞数据注释方法的流程示意图;
22.图3为本技术实施例提供的细胞数据注释的结构示意图;
23.图4为本技术实施例提供的获得初始细胞注释结果方法的流程示意图;
24.图5为本技术实施例提供的获得细胞注释结果方法的流程示意图;
25.图6为本技术实施例提供的通过第一预测模型得到输出结果的结构示意图;
26.图7为本技术另一实施例提供的通过第一预测模型得到输出结果的结构示意图;
27.图8为本技术又一实施例提供的通过第一预测模型得到输出结果的结构示意图;
28.图9为本技术实施例提供的细胞数据注释方法的流程示意图;
29.图10为本技术实施例提供的对第一预测模型进行训练方法的流程示意图;
30.图11为本技术实施例提供的对第一预测模型进行训练方法的流程示意图;
31.图12为本技术实施例提供的对待预测转录组进行预测方法的流程示意图;
32.图13为本技术实施例提供的细胞数据注释装置的结构示意图;
33.图14为本技术另一实施例提供的细胞数据注释装置的结构示意图;
34.图15为本技术实施例示出的一种计算机设备的结构示意图。
具体实施方式
35.下面结合附图和实施例对本技术作进一步的详细说明。可以理解的是,此处所描述的具体实施例仅仅用于解释相关发明,而非对该发明的限定。另外还需要说明的是,为了便于描述,附图中仅示出了与发明相关的部分。
36.需要说明的是,在不冲突的情况下,本技术中的实施例及实施例中的特征可以相互组合。下面将参考附图并结合实施例来详细说明本技术。为了便于理解,下面对本技术实施例涉及的一些技术术语进行解释:
37.(1)人工智能(artificial intelligence,ai):是利用数字计算机或者数字计算机控制的机器模拟、延伸和扩展人的智能,感知环境、获取知识并使用知识获得最佳结果的理论、方法、技术及应用系统。换句话说,人工智能是计算机科学的一个综合技术,它企图了解智能的实质,并生产出一种新的能以人类智能相似的方式做出反应的智能机器。人工智能也就是研究各种智能机器的设计原理与实现方法,使机器具有感知、推理与决策的功能。
38.人工智能技术是一门综合学科,涉及领域广泛,既有硬件层面的技术也有软件层面的技术。人工智能基础技术一般包括如传感器、专用人工智能芯片、云计算、分布式存储、大数据处理技术、操作/交互系统、机电一体化等技术。人工智能软件主要包括计算机视觉、语音处理技术、自然语言技术以及机器学习/深度学习等几大方向。
39.(2)机器学习(machine learning,ml):是一门多领域交叉学科,涉及概率论、统计学、逼近论、凸分析、算法复杂度理论等多门学科。专门研究计算机怎么模拟或实现人类的学习行为,以获取新的知识或技能,重新组织已有的知识结构使之不断改善自身的性能。机器学习使人工智能的核心,是使计算机具有智能的根本途径,其应用遍及人工智能的各个领域。机器学习和深度学习通常包括人工神经网络、置信网络、强化学习、迁移学习、归纳学习、示教学习等技术。
40.(3)深度神经网络(deep neural networks,ddn):是一种具备至少一个隐层的神经网络。与浅层神经网络类似,深度神经网络能够为复杂非线性系统提供建模,但多出的层次为模型提供了更高的抽象层次,因此提高了模型的能力。
41.(4)空间转录组(spatial transcriptomics,st):可以是某一生理条件下,细胞内所有转录产物的集合,例如,可以是信使rna、核糖体rna、转运rna及其他非编码rna。或者,也可以是认为空间转录组是所有mrna的集合。
42.(5)细胞注释结果:是指对空间转录组数据进行分析和细胞注释后得到的处理结果,用于标识空间转录组中细胞数据的身份信息,以便能够快速获取空间转录组数据的细胞类型信息和细胞属性特点等。
43.随着人工智能技术研究和进步,人工智能技术在多个领域展开研究和应用,例如常见的智能家居、智能穿戴社保、虚拟助理、智能音箱、智能营销、无人驾驶、自动驾驶、无人机、机器人、智能医疗、智能客服等,相信随着技术的发展,人工智能技术将在更多的领域得到应用,并发挥越来越重要的价值。
44.本技术实施例提供的方案涉及人工智能的神经网络等技术,具体通过下述实施例进行说明。
45.目前,相关技术中可以采用分类训练的方式基于参考数据集构建模型,然后通过构建的模型预测待注释数据集的细胞类型。但是该自动注释方法在预测细胞类型的过程
中,利用参考数据集的比较单一片面,导致对细胞数据注释的准确度较低。
46.基于上述缺陷,本技术提供了一种细胞数据注释方法、装置、设备及介质,与现有技术相比,能够基于特征较为全面的细胞数据获得待预测转录组的细胞注释结果,从而提高了空间转录组的细胞注释结果的准确性。
47.图1是本技术实施例提供的一种细胞数据注释方法的实施环境架构图。如图1所示,该实施环境架构包括:终端10和服务器20。
48.其中,在测序分析领域,对空间转录组的细胞数据进行注释的过程即可以在终端10执行,也可以在服务器20执行。例如,通过终端10接收用户输入的细胞数据,可以在终端10本地进行细胞注释,得到细胞注释结果;也可以将细胞数据发送至服务器20,使得服务器20接收细胞数据,根据细胞数据进行细胞注释,得到细胞注释结果,然后将细胞注释结果发送至服务器20,以实现对待预测转录组的细胞数据的细胞注释。
49.本技术实施例提供的对空间转录组细胞数据注释方案,提高了计算机设备对空间转录组细胞注释的准确率,上述细胞注释结果可以应用于终端或服务器等计算机设备涉及的多个测序分析领域,例如,基因差异表达分析、细胞发育轨迹分析、基因本体(gene ontology,go)富集分析等。也可作为初步的细胞注释结果提供给用户进行修改和微调,以获得用户认为更加可信的完整细胞注释结果。
50.另外,终端10可显示有应用界面,通过该界面可获取用户上传的待预测转录组的细胞数据,或将上传的待预测转录组的细胞数据发送给服务器20。
51.可选的,终端10可以是各类ai应用场景中的终端设备。例如,终端10可以是智能电视、智能手机、平板电脑、电视机、笔记本电脑、台式电脑等,本技术实施例对此不进行具体限定。
52.服务器20可以是一台服务器,也可以是由若干台服务器构成的服务器集群或者分布式系统,还可以是提供云服务、云数据库、云计算、云函数、云存储、网络服务、云通信、中间件服务、域名服务、安全服务、内容分发网络(content delivery network,cdn)、以及大数据和人工智能平台等基础云计算服务的云服务器。
53.终端10与服务器20之间通过有线或无线网络建立通信连接。可选的,上述的无线网络或有线网络使用标准通信技术和/或协议。网络通常为因特网、但也可以是任何网络,包括但不限于局域网(local area network,lan)、城域网(metropolitan area network,man)、广域网(wide area network,wan)、移动、有线或者无线网络、专用网络或者虚拟专用网络的任何组合。
54.为了便于理解和说明,下面通过图2至图15详细阐述本技术实施例提供的细胞数据注释方法、装置、设备及介质。
55.图2所示为本技术实施例的细胞数据注释方法的流程示意图,该方法可以由计算机设备执行,该计算机设备可以是上述图1所示系统中的服务器20或者终端10,或者,该计算机设备也可以是终端10和服务器20的结合。如图2所示,该方法包括:
56.s101、获取待预测转录组的细胞数据;细胞数据包括待预测转录组中多个测序点的基因表达信息以及多个测序点的空间信息。
57.上述待预测转录组的细胞数据可以是对待预测样本(组织或部位)进行基因测序获得的信息。例如,可以在待预测样本的切片上选择测序点,并对测序点进行基因测序获得
待预测转录组的细胞数据。其中,上述组织可以是脑部组织、心脏组织以及肺部组织等。
58.需要说明的是,上述测序点可以是切片中用于获取待预测转录组的转录本信息的一些细胞。
59.在一种可能的实现方式中,对测序点进行基因测序获得的信息可以包括测序点的基因表达信息以及测序点的空间信息。基因表达信息用于表征各个测序点的基因信息,测序点的空间信息用于表征各个测序点的空间位置信息。也就是说,转录组的细胞数据可以包括转录组中各个测序点的空间信息以及各个测序点的基因表达信息。
60.一种可能的实现方式中,上述空间转录组的细胞数据的获取方式可以包括:(1)结合显微成像技术和测序技术获取某一组织后切片中的转录组数据,然后调用10x genomics visium平台对数据进行分析处理和可视化获得;
61.(2)基于转录组测序以及基于激光显微切割(laser capture microdissection,lcm)的转录组分析方法获得,该测序方法例如可以包括新的空间转录组方法(transcriptome in vivo analysis,tiva),锁式探针原位测序的分析方法(in situ sequencing,iss),荧光原位测序(fluorescence in situ sequencing,fisseq);
62.(3)通过外部设备直接导入获取得到。
63.s102、确定与待预测转录组对应的已注释细胞对象,根据细胞对象以及基因表达信息确定待预测转录组的初始细胞注释结果。
64.具体地,上述待预测转录组对应的已注释细胞对象可以是与所述待预测转录组同源、且细胞注释结果为已知的细胞对象。例如,已注释细胞对象可以是待预测转录组对应的基因测序对象包括的单细胞或细胞组。其中,单细胞是指每个样本是以细胞为单位进行测序形成的细胞数据。细胞组是指每个样本是多个细胞为单位进行测序形成的细胞数据,例如bulk数据,还可以每个样本是亚细胞级的细胞数据。
65.需要说明的是,待预测转录组对应的基因测序对象可以是待预测转录组同源的基因测序对象,例如,与待预测转录组取自相同组织或部位的基因测序对象。示例性的,取自脑部的待预测转录组与取自脑部的基因测序对象是对应的,而与取自心脏的基因测序对象不对应。取自心脏的待预测转录组的细胞数据与取自心脏的基因测序对象对应。其中,若待预测转录组的细胞数据为大脑切片,则确定的基因测序对象也应该取自大脑同一区域;若待预测转录组的细胞数据为心脏切片,则确定出对应的基因测序对象也应取自心脏同一区域。
66.本实施例中通过确定与待预测转录组对应的细胞对象,能够使得针对正确的细胞对象确定获取待预测转录组细胞注释结果的指导信息,进而使得确定出的初始细胞注释结果更准确。
67.本技术实施例中,在确定与待预测转录组对应的已注释细胞对象的过程中,作为一种可能的实现方式,可以是通过对已注释细胞数据进行识别,判断细胞数据的区域或组织是否与待预测转录组的细胞数据对应的区域或组织相同。一种可能的实现方式中,可以通过观察大量数据集中细胞的形态和结构特征,根据细胞的形态和结构特征确定细胞对象,例如,上皮细胞由密集细胞和细胞间质组成,肌肉组织由肌细胞组成且呈纤维状,神经组织由神经元和神经胶质细胞组成。另一种可能的实现方式中,可以从预设细胞数据库中查询并获取与待预测转录组取自相同组织或区域的已注释细胞数据,将该已注释细胞数据
确定为与待预测转录组对应的已注释细胞对象。其中,该预设细胞数据库可以是对不同细胞类型、细胞形态和结构等特征的细胞数据进行汇总和分类整理后构建的。
68.进一步地,在根据细胞对象以及基因表达信息确定待预测转录组的初始细胞注释结果时,作为一种可实现方式,可以采用单细胞迁移的方式,根据单细胞以及待预测转录组中的基因表达信息确定待预测转录组的初始细胞注释结果,例如可以预先基于单细胞和与单细胞对应的细胞注释结果训练得到神经网络模型,该神经网络模型具有对待预测转录组的细胞数据进行细胞类型注释的能力,然后通过该神经网络模型对待预测转录组的细胞数据进行预测,得到初始细胞注释结果。
69.作为另一种可实现方式,可以采用其他非单细胞迁移的方式,例如非单细胞为每个样本是以多个细胞时,则对应算法可以是结合反卷积和迁移的方式,以得到待预测转录组的初始细胞注释结果;例如非单细胞为亚细胞级时,对应算法是结合聚合和迁移的方式,以得到待预测转录组的初始细胞注释结果。
70.还提供一种可能的实现方式,还可以是通过细胞类型特异性表达的mark标志基因识别的方式,对基因表达信息和空间信息进行综合分析以确定待预测转录组的初始细胞注释结果;也可以采用引入相应领域的先验知识的方式,通过聚类算法对待预测转录组的细胞数据进行聚类,得到聚类结果,然后利用人工先验生物学知识确定待预测转录组中每个聚类结果的初始细胞注释结果;还可以查询预先建立的已知细胞类型的转录谱数据库,将未知类型的待预测转录组的细胞数据的细胞特征与已知细胞类型的转录谱数据库的细胞特征进行比对,将细胞特征相同的细胞类型确定为待预测转录组的初始细胞注释结果。
71.需要说明的是,上述根据细胞对象以及基因表达信息确定待预测转录组的初始细胞注释结果的各个实现方式仅仅是作为一种示例,本技术实施例对此不做限定。
72.本技术实施例中在确定与待预测转录组对应的细胞对象后,根据细胞对象和基因表达信息确定待预测转录组的初始细胞注释结果,能够为最终细胞注释结果的确定提供了指导信息,以便为最终细胞注释结果提供了准确的数据依据,进而提高了细胞数据注释的准确度。
73.s103、将细胞数据输入第一预测模型,根据第一预测模型的输出与初始细胞注释结果之间的损失对初始细胞注释结果进行修正,获得待预测转录组的细胞注释结果。
74.需要说明的是,上述第一预测模型是一个输入为待预测转录组的基因表达信息和空间信息,输出为待预测转录组的细胞注释结果,且具有对待预测转录组的基因表达信息和空间信息进行信息整合的能力,能够预测细胞注释结果的神经网络模型。该第一预测模型可以是通过自监督算法进行迭代训练时的初始模型,即第一预测模型的模型参数处于初始的状态,也可以是上一轮迭代训练中调整后的模型,即第一预测模型的模型参数处于中间的状态。可以将细胞数据输入第一预测模型,得到输出结果。该输出结果包括待预测转录组的细胞类型,或者可以包括待预测转录组在细胞类型下的多个细胞属性。
75.具体地,请参见图3所示,在确定出待预测转录组对应的细胞对象后,可以根据细胞对象和基因表达信息确定待预测转录组的初始细胞注释结果310。例如,可以根据上述已注释细胞对象的细胞注释方式,来获取待预测转录组的初始细胞注释结果。
76.可选地,在将细胞数据输入第一预测模型320时,可以是通过融合基因表达信息和空间信息,得到融合特征信息,然后将融合特征信息进行分类处理,得到第一预测模型的输
出结果330,即第一预测模型预测的上述待预测转录组的细胞注释结果。还可以根据第一预测模型的输出结果330与初始细胞注释结果310之间的损失对初始细胞注释结果310进行修正,获得待预测转录组的细胞注释结果340。具体地,根据第一预测模型的输出与初始细胞注释结果之间的损失对初始细胞注释结果进行修正的过程,可以是将初始细胞注释结果作为伪标签,利用图神经网络(graph convolutional network,gcn)的网络框架整合待预测转录组的基因表达信息和空间信息,并通过分类模型进行分类得到第一预测模型的输出结果,然后根据输出结果以自监督训练的方式对初始细胞注释结果进行修正,从而得到调整后的第一预测模型,进而将细胞数据通过调整后的第一预测模型进行预测处理,从而获取待预测转录组的细胞注释结果。可选的,上述第一预测模型可以包括编码-解码模块和分类模块,该编码-解码模块例如可以是深度自编码器(deep auto-encoder,dae)和变分图自编码器(variational graph auto-encoder,vgae),还可以是其他自编码模块,如vgae可替换为图神经网络graphsage等其他图网络框架。其中,graphsage相当于一种聚合相邻节点信息的框架,在这个框架下,可以使用不同的聚合函数来结合相邻节点的信息和来自当前节点上一层的信息,其中,聚合函数可以用mean、pool、lstm等。
77.其中,上述细胞注释结果用于标识待预测转录组,以便能够通过待预测转录组的注释结果快速获取到待预测转录组的信息、特点等。例如,待预测转录组的注释结果可以包括待预测转录组的细胞类型,或者可以包括待预测转录组在细胞类型下的多个细胞属性。例如,细胞类型可以是上皮细胞、t细胞、纤维细胞、形状胶质细胞和内皮细胞等。其中,t细胞对应的细胞属性例如可以是辅助性t细胞、抑制性t细胞、效应t细胞、细胞毒性t细胞、记忆t细胞等。不同细胞属性对应的t细胞的功能不同。辅助性t细胞具有协助体液免疫和细胞免疫的功能;抑制性t细胞具有抑制细胞免疫及体液免疫的功能;效应t细胞具有释放淋巴因子的功能;细胞毒性t细胞具有杀伤靶细胞的功能;记忆t细胞具有记忆特异性抗原刺激的功能。
78.在对待预测转录组进行细胞数据注释的过程中,作为一种可实现方式,在对待预测转录组进行细胞数据注释时,可以获取待预测转录组的细胞数据,该细胞数据包括待预测转录组中多个测序点的基因表达信息以及多个测序点的空间信息,然后确定与待预测转录组对应的已注释细胞对象,通过对从大量数据集中获取细胞数据取自的组织或区域,然后将取自的组织或区域与待预测转录取自的组织或区域相同的细胞数据确定为细胞对象,该细胞对象可以是待预测转录组对应的单细胞或细胞组。并根据细胞对象和基因表达信息确定待预测转录组的初始细胞注释结果,可以通过单细胞迁移的方式,根据单细胞以及待预测转录组中的基因表达信息确定待预测转录组的初始细胞注释结果。
79.示例性地,例如预先基于单细胞和单细胞对应的细胞注释结果训练得到神经网络模型,然后将待预测转录组的细胞数据输入该神经网络模型中,经过预测处理得到初始细胞注释结果,可以将该初始细胞注释结果作为待预测转录组的最终的细胞注释结果。该实施例中仅仅是利用已知注释结果的单细胞数据对待预测转录组的细胞数据进行注释,未能捕捉到单细胞和空间数据的测序差异,且没有利用到最重要的空间信息,从而得到的细胞注释结果精准度较差。
80.作为另一种可实现方式,可以将单细胞迁移的方式或其他单细胞迁移的方式,根据待预测转录组的基因表达信息确定初始细胞注释结果,并将初始细胞注释结果作为自监
督模型的伪标签,然后将该待预测转录组的细胞数据输入第一预测模型,得到输出结果,并根据第一预测模型的输出结果与伪标签之间的损失对初始细胞注释结果进行修正,从而得到待预测转录组的细胞注释结果。
81.本技术实施例提供的细胞数据注释方法中,通过确定与待预测转录组对应的细胞对象,并根据细胞对象以及基因表达信息确定待预测转录组的初始细胞注释结果,其在无需任何人工参与注释的情况下,能够获取到待预测转录组的细胞注释结果的指导信息,并且通过第一预测模型对细胞数据进行预测,根据第一预测模型的输出与初始细胞注释结果之间的损失对初始细胞注释结果进行修正,有效地融合了多个测序点的基因表达信息以及多个测序点的空间信息,能够结合指导信息进行自监督修正,从而更为精准、全面地利用待预测转录组的细胞数据中的所有信息,进而更加准确地获得待预测转录组的细胞注释结果,很大程度上提升了对待预测转录组的细胞数据注释的准确度。还可以应用于测序分析系统中,对待预测转录组的细胞数据进行准确的预测,极大地提升了细胞注释的质量和效率,为空间转录组的数据分析提供了强有力的支持。
82.在本技术的另一实施例中,可以借助已注释细胞对象所适用的神经网络模型来获得待预测转录组的初始细胞注释结果。图4提供了确定待预测转录组的初始细胞注释结果的具体实现方式。请参见图4所示,具体包括:
83.s201、确定与细胞对象匹配的第二预测模型;第二预测模型是基于细胞对象的历史基因表达信息以及历史基因表达信息对应的历史细胞注释结果进行训练获得的。
84.具体地,上述与已注释细胞对象匹配的第二预测模型,即能够对已注释细胞对象进行注释结果预测的神经网络模型。该第二预测模型是基于细胞对象的历史基因表达信息以及历史基因表达信息对应的历史细胞注释结果进行训练获得的。其中,可以是细胞对象的真实细胞类型或真实细胞属性。该第二预测模型可以基于基因表达信息获得初始细胞注释结果。可以先通过提取基因表达信息中的特征信息,然后将该特征信息通过全连接层得到全连接向量,最后将该全连接向量通过激活函数进行处理,从而得到初始细胞注释结果。
85.一种可能的实现方式中,确定与细胞对象匹配的第二预测模型,即在执行步骤s201时获取上述第二预测模型的模型文件,以便后续运行该模型文件对待预测转录组的注释结果进行预测。
86.另一种可能的实现方式中,可以在执行步骤s201时获取已注释细胞对象的历史基因表达信息以及历史基因表达信息对应的历史细胞注释结果,然后根据该历史基因表达信息以及历史基因表达信息对应的历史细胞注释结果进行实时训练获得的上述第二预测模型。
87.其中,细胞对象可以是单细胞或细胞组。该第二预测模型具有对细胞数据进行细胞类型注释的能力,该第二预测模型可以是通过预设训练算法进行训练得到的,即第二预测模型的模型参数已处于最优的状态。历史细胞注释结果可以是细胞对象的真实细胞类型或真实细胞属性。
88.可选的,上述神经网络模型可以是深度神经网络(deep neural networks,dnn)模型、卷积神经网络(convolutional neural networks,cnn)模型、长短期记忆神经元(long short term memory cells,lstm)模型或循环神经网络(recurrent neural networks,rnn)模型等。
89.一种可能的实现方式中,上述第二预测模型的训练过程具体包括:将历史基因表达信息按照一定比例随机分为训练集和验证集,然后利用训练集和验证集按照训练学习算法构建得到第二预测模型。其中,训练集用于对初始第二预测模型进行训练,以得到训练好的第二预测模型,验证集用于对训练好的第二预测模型进行验证,以验证第二预测模型性能的好坏。
90.其中,计算设备在训练第二预测模型的过程中,利用验证集中对待验证的第二预测模型,按照损失函数最小化对待验证的第二预测模型进行优化处理,得到第二预测模型,根据该验证集输入待验证的第二预测模型中得到的结果和历史基因表达信息对应的历史细胞注释结果之间的差异,对待构建的第二预测模型中的参数进行更新,以实现对第二预测模型进行训练的目的,其中,上述历史细胞注释结果可以是人工对历史基因表达信息进行标注得到的结果。
91.本技术实施例中,在训练第二预测模型时,可以使用损失函数计算验证集输入待验证的第二预测模型中得到的结果和历史细胞注释结果的损失值,从而对待验证的第二预测模型中的参数进行更新。可选的,损失函数可以使用交叉熵损失函数,归一化交叉熵损失函数,或者可以使用focalloss等。
92.s202、将基因表达信息输入第二预测模型,获得待预测转录组的初始细胞注释结果。
93.在构建得到第二预测模型后,可以将待预测转录组的基因表达信息输入第二预测模型中进行预测处理,得到待预测转录组的初始细胞注释结果。其中,初始细胞注释结果可以包括待预测转录组的细胞类型,或者可以包括待预测转录组在细胞类型下的多个细胞属性。
94.一种可能的实现方式中,上述第二预测模型对基因表达信息的处理具体包括:在对基因表达信息进行处理的过程中,可以是先通过对待预测转录组进行特征提取,得到特征信息,再通过多分类函数对特征信息进行运算,输出细胞类型。还可以通过多元二分类对特征信息进行运算,输出细胞属性。可选的,上述多分类函数可以是softmax函数,上述多元二分类函数可以是多个sigmoid函数,一个sigmoid函数可以实现一个二分类预测。其中,上述多分类函数的作用是用来加入非线性因素,因为线性模型的表达能力不够,能够把输入的连续实值变换为0和1之间的输出。
95.示例性地,将基因表达信息输入第二预测模型中,第二预测模型的预测结果可以包括“t细胞”、“上皮细胞”和“纤维细胞”等细胞类型中的任意一个。其中,预测结果还可以包括细胞属性,例如细胞类型“t细胞”对应的细胞属性可以是“辅助性t细胞”、“抑制性t细胞”、“效应t细胞”、“细胞毒性t细胞”、“记忆t细胞”中的多个。
96.其中,以三分类为例,介绍多分类函数的输出。例如,多分类函数能够预测的细胞类型分别为“t细胞”、“上皮细胞”和“纤维细胞”,上述神经网络模型的输出结果可以是通过向量表示,例如可以是一个3*1维的向量,该向量中的每一个元素对应一个细胞类型,向量中的每个元素值表示待预测转录组为对应标签种类的概率。假设多分类函数的输出向量为[0.61,0.31,0.08],则表示待预测转录组为“t细胞”的概率为0.61,待预测转录组为“上皮细胞”的概率为0.31,待预测转录组为“纤维细胞”的概率为0.08,可以选择概率最大的元素值作为待预测转录组的预测结果,即将“t细胞”作为待预测转录组的初始细胞注释结果。
[0097]
以三元二分类为例,介绍多元二分类函数的输出。例如,多元二分类函数能够预测的细胞属性用“辅助性t细胞”、“抑制性t细胞”、“效应t细胞”和“记忆t细胞”表示,其输出结果可以是通过向量表示,例如可以是一个4*1维的向量,该向量中的每一个元素对应一个细胞属性,向量中的每个元素值表示待预测转录组为对应细胞属性的概率。假设三元二分类函数的输出向量为[0.51,0.15,0.22、0.62],则表示这个待预测转录组为“辅助性t细胞”的概率为0.51,待预测转录组为“抑制性t细胞”的概率为0.15,待预测转录组为“效应t细胞”的概率为0.62,待预测转录组为“记忆t细胞”的概率为0.12。假设预设阈值为0.5,将概率大于预设阈值的概率的元素值为作为待预测转录组的预测结果,即将“辅助性t细胞”和“效应t细胞”作为多元二分类函数的初始细胞注释结果。
[0098]
本技术实施例中,通过将待预测转录组输入第二预测模型进行预测处理,很大程度上提高了确定细胞注释结果的准确度,能够更加精准地得到初始细胞注释结果,为最终细胞注释结果的确定提供了精准的指导信息,实现更高精度的细胞数据注释。
[0099]
在本技术的另一实施例中,还提供了基于第一预测模型对初始细胞注释结果进行修正的具体实现方式。例如,图5提供了获取待预测转录组的细胞注释结果的具体实现方式。请参见图5所示,该方法包括:
[0100]
s301、将细胞数据输入第一预测模型进行编码处理得到融合编码信息,将融合编码信息输入第一预测模型的分类模块,获得第一预测模型的输出结果。
[0101]
具体的,该待注释转录组的细胞数据可以包括待预测转录组中多个测序点的基因表达信息,以及多个测序点的空间信息,其中,该基因表达信息、空间信息可以为矩阵或向量。请参见图6所示,将待预测转录组输入第一预测模型进行编码处理,可以包括:将细胞数据输入第一预测模型中的编码模块410基于基因表达信息和空间信息进行信息融合、编码,从而提取到融合特征信息,得到融合编码信息,该融合编码信息的表现形式可以为矩阵或向量形式,其中,该编码模块可以包括第一编码模块和第二编码模块。
[0102]
将融合编码信息输入第一预测模型的分类模块420,得到第一预测模型的输出结果430。该分类模块用于负责建立融合特征信息与目标细胞类型之间的关系。其中,该分类模块可以包括但不限于全连接层和激活函数,全连接层可以包括一层,或者也可以包括多层。全连接层主要是用于对融合特征信息进行分类的作用。可以将融合特征信息通过全连接层进行处理,得到全连接向量,然后将全连接向量通过激活函数进行处理,从而得到第一预测模型的输出结果,该输出结果可以为细胞类型。
[0103]
其中,上述激活函数可以是sigmoid函数,也可以是tanh函数,还可以是relu函数,通过将全连接向量经过激活函数处理,能够将其结果映射到0~1之间。
[0104]
作为一种可选的实施方式,提供了上述编码模块的具体处理流程。请参见图7所示,在将细胞数据输入第一预测模型进行编码处理的过程中,可以是将基因表达信息输入第一预测模型的第一编码模块510,获得第一编码信息,然后根据空间信息确定邻接矩阵,该邻接矩阵用于表征各个测序点之间相邻关系的矩阵,包括待预测转录组中各个测序点的邻近测序点。并将邻接矩阵和第一编码信息输入第一预测模型的第二编码模块520进行处理,得到第二编码信息,最后将第一编码信息和第二编码信息进行融合处理,得到融合编码信息。然后将融合编码信息输入分类模块530中进行分类处理,得到输出结果540。
[0105]
其中,上述第一编码模块用于对基因表达信息进行特征编码,其可以是深度自编
码器dae,第二编码模块用于对空间信息进行特征编码。第一编码信息是指对基因表达信息进行特征编码后得到的编码信息,其可以是变分图自编码器vgae。第二编码信息是指对基因表达信息和空间信息进行编码后得到的编码信息。上述第一编码信息和第二编码信息可以通过向量或矩阵表示。
[0106]
需要说明的是,在将第一编码信息和第二编码信息进行融合处理的过程中,当第一编码信息和第二编码信息为通过向量表示时,可以是通过向量组合的方式进行信息融合;当第一编码信息和第二编码信息为通过矩阵表示时,可以是通过矩阵拼接的方式进行信息融合,从而得到对应的融合编码信息。
[0107]
s302、根据第一预测模型的损失函数确定输出结果和初始细胞注释结果之间的损失,根据损失迭代训练分类模块,在损失函数最小化时获得调整后的第一预测模型。
[0108]
可选的,上述第一预测模型的损失函数可以为分类损失,例如为初始细胞注释结果与第一预测模型输出之间的损失;也可以为重构损失,例如为重构的基因表达信息和第一预测模型输入的基因表达信息之间的损失,还可以为重构的空间信息和第一预测模型输入的空间信息之间的损失;也可以包括分类损失与重构损失,其中包括重构损失中的一个或两个与分类损失之和。
[0109]
在得到输出结果后,可以根据损失函数确定输出结果与初始细胞注释结果之间的损失,根据损失迭代训练分类模块,在损失函数最小化时获得调整后的第一预测模型,例如可以采用梯度下降法进行迭代训练。其中,在根据损失迭代训练第一预测模型时,可以通过对分类模块中的参数进行更新,例如可以是对分类模块中的权重矩阵以及偏置矩阵等矩阵参数进行更新。其中,上述权重矩阵、偏置矩阵包括但不限于是分类模块的自注意力层、前馈网络层、全连接层中的矩阵参数。
[0110]
其中,通过损失函数对第一预测模型中的参数进行更新时,可以是根据损失函数确定第一预测模型未收敛时,通过调整第一预测模型中的参数,可以是通过调整分类模块中的参数,以使得第一预测模型收敛,从而得到调整后的第一预测模型。其中,第一预测模型收敛,可以是指第一预测模型的输出结果与初始细胞注释结果小于预设阈值,或者,输出结果与初始细胞注释结果之间的差值的变化率趋近于某一个较低值。当计算的损失函数较小,或者,与上一轮迭代输出的损失函数之间的差值趋近于0,则认为分类模块收敛,进而认为第一预测模型收敛。
[0111]
s303、将细胞数据输入调整后的第一预测模型,获得待预测转录组的细胞注释结果。
[0112]
需要说明的是,上述调整后的第一预测模型可以是通过训练算法迭代训练得到的模型,即调整后的第一预测模型的模型参数已处于最优的状态,其中,该训练算法可以是自监督学习算法。可选的,该调整后的第一预测模型可以包括调整后的编码模块和分类模块。其中,该调整后的编码模块包括第三编码模块和第四编码模块。第三编码模块用于对基因表达信息进行特征编码,第四编码模块用于对空间信息进行特征编码。
[0113]
具体地,在得到调整后的第一预测模型后,可以将待预测转录组的细胞数据输入调整后的第一预测模型,该细胞数据包括待预测转录组的基因表达信息和空间信息,可以将基因表达信息输入调整后的第一预测模型中的第三编码模块,获得第三编码信息,然后根据空间信息确定邻接矩阵,并将邻接矩阵和第三编码信息输入调整后的第一预测模型的
第四编码模块,获得第四编码信息,并将第三编码信息和第四编码信息进行融合处理,得到合并编码信息。将合并编码信息输入调整后的第一预测模型的分类模块进行分类处理,得到待预测转录组的细胞注释结果。
[0114]
本实施例中通过将细胞数据输入第一预测模型进行编码处理和分类处理得到输出结果,并根据输出结果和初始注释结果之间的损失,迭代训练获得调整后的第一预测模型,能够通过自监督训练的方式有效地提取大基因表达信息和空间信息,使得构建出的调整后第一预测模型更优,进而使得获取的细胞注释结果的准确度更高。
[0115]
在本技术的另一实施例中,还可以对第一预测模型中的第一解码模块进行迭代训练。具体地,在得到融合编码信息之后,还可以对融合编码信息进行解码处理,获得重构的基因表达信息,并根据损失函数确定重构的基因表达信息与基因表达信息之间的损失,根据损失调整第一编码模块的参数,使得重构的基因表达信息接近基因表达信息。
[0116]
具体地,请参见图8所示,在对融合编码信息进行解码处理的过程中,可以是先将融合编码信息进行线性特征提取处理,得到第一中间特征信息,通过线性层进行特征提取处理,然后将第一中间特征信息输入第一解码模块610进行特征还原处理,获取重构的基因表达信息。其中,上述线性层的数量可以是两个或多个,线性层的数量越多,则从融合编码信息中进行特征提取后得到的特征信息越准确。该第一中间特征信息可以为线性处理后的基因表达信息。
[0117]
需要说明的是,上述第一解码模块的作用是将重构的基因表达信息能够准确地还原回原始输入的基因表达信息。
[0118]
进一步地,在得到重构的基因表达信息后,可以根据损失函数确定重构的基因表达信息与基因表达信息之间的损失,根据损失迭代训练第一编码模块,在损失函数最小化时获得调整后的第一预测模型,例如可以采用梯度下降法进行迭代训练。
[0119]
其中,在根据损失迭代训练第一预测模型时,可以通过对第一编码模块中的参数进行更新,例如可以是对第一编码模块中的权重矩阵以及偏置矩阵等矩阵参数进行更新。其中,上述权重矩阵、偏置矩阵包括但不限于是第一编码模块的输入层、隐藏层中的矩阵参数。
[0120]
其中,通过损失函数对预测模块中的参数进行更新时,可以是根据损失函数确定预测模块未收敛时,通过调整第一编码模块中的参数,可以是通过调整第一编码模块中的参数,以使得第一预测模型收敛,从而得到调整后的第一预测模型。其中,第一预测模型收敛,可以是指重构的基因表达信息与第一预测模型输入的基因表达信息小于预设阈值,或者,重构的基因表达信息与第一预测模型输入的基因表达信息之间的差值的变化率趋近于某一个较低值。当计算的损失函数较小,或者,与上一轮迭代输出的损失函数之间的差值趋近于0,则认为第一编码模块收敛,进而认为第一预测模型收敛。
[0121]
在本技术的另一实施例中,还可以对第一预测模型中的第二解码模块进行迭代训练。具体地,在得到融合编码信息之后,还可以对融合编码信息进行解码处理,获得重构的空间信息,并根据损失函数确定重构的空间信息与空间信息之间的损失,根据损失调整第二编码模块的参数,使得重构的空间信息接近空间信息。
[0122]
具体地,在对融合编码信息进行解码处理的过程中,可以是先将融合编码信息进行线性特征提取处理,得到对应的第二中间特征信息,通过线性层进行特征提取处理,然后
将第二中间特征信息输入第二解码模块620进行特征还原处理,获取重构的空间信息。其中,上述线性层的数量可以是两个或多个,线性层的数量越多,则从融合编码信息中进行特征提取后得到的特征信息越准确。该第二中间特征信息可以为线性处理后的空间信息。
[0123]
需要说明的是,上述第二解码模块的作用是将重构的空间信息能够准确地还原回原始输入的空间信息。
[0124]
进一步地,在得到重构的空间信息后,可以根据损失函数确定重构的空间信息与空间信息之间的损失,根据损失迭代训练第二编码模块,在损失函数最小化时获得调整后的第一预测模型,例如可以采用梯度下降法进行迭代训练。
[0125]
其中,在根据损失迭代训练第一预测模型时,可以通过对第二编码模块中的参数进行更新,例如可以是对第二编码模块中的权重矩阵以及偏置矩阵等矩阵参数进行更新。其中,上述权重矩阵、偏置矩阵包括但不限于是第二编码模块的输入层、隐藏层中的矩阵参数。
[0126]
其中,通过损失函数对预测模块中的参数进行更新时,可以是根据损失函数确定预测模块未收敛时,通过调整第二编码模块中的参数,可以是通过调整第二编码模块中的参数,以使得第一预测模型收敛,从而得到调整后的第一预测模型。其中,第一预测模型收敛,可以是指重构的空间信息与第一预测模型输入的空间信息小于预设阈值,或者,重构的空间信息与第一预测模型输入的空间信息之间的差值的变化率趋近于某一个较低值。当计算的损失函数较小,或者,与上一轮迭代输出的损失函数之间的差值趋近于0,则认为第二编码模块收敛,进而认为第一预测模型收敛。
[0127]
本实施例中通过对融合编码信息进行解码处理,得到重构后的基因表达信息和重构后的空间信息,能够结合基因表达信息对第一编码器和第二编码器中的参数进行精准地调整,进而使得调整后的第一预测模型准确度更高,进而提高确定细胞注释结果的准确度。
[0128]
需要说明的是,本技术实施例第一编码模块、第二编码模块的迭代训练是两个独立的处理过程,可以仅进行第一编码模块的迭代训练,也可以仅进行第二编码模块的迭代训练。当然,也可以对第一编码模块、第二编码模块均进行迭代训练,对二者的执行顺序不作限定,可以是在一次迭代训练中串联执行,也可以是并行执行。以并行执行为例,在得到融合编码信息之后,还可以对融合编码信息进行解码处理,获得重构的空间信息和重构的基因表达信息,并根据损失函数确定重构的空间信息与空间信息之间的损失,以及确定重构的基因表达信息与基因表达信息之间的损失,根据损失同时调整第一编码模块和第二编码模块的参数,使得重构的空间信息接近空间信息,以及使得重构的基因表达信息接近基因表达信息。
[0129]
在本技术的另一实施例中,还提供了第一预测模型的损失函数的具体实现。一种可能的实现方式中,第一预测模型的损失函数包括第一分量、第二分量和第三分量。第一分量用于表征初始细胞注释结果与第一预测模型的输出之间的损失;第二分量用于表征重构的基因表达信息与第一预测模型的输入中的基因表达信息之间的损失;重构的基因表达信息是对融合编码信息进行解码处理后获得的;第三分量用于表征重构的空间信息与第一预测模型的输入中的空间信息之间的损失;重构的空间信息是对融合编码信息进行解码处理后获得的。
[0130]
其中,上述构建的损失函数可以是第一分量、第二分量和第三分量之和。通过在损
失函数中设置第二分量和第三分量,能够缩小重构的基因表达信息和第一预测模型输入的基因表达信息,以及重构的空间信息和第一预测模型输入的空间信息之间的差异,从而保证了融合特征信息融合了待预测转录组的基因表达信息和空间信息,且通过设置第一分量,能够对融合特征信息得到最终细胞类型提供指导信息,使得得到的调整后的第一预测模型达到更优。
[0131]
本技术实施例中,在构建损失函数以得到调整后的第一预测模型时,综合了初始细胞注释结果与第一预测模型的输出之间的差异,和重构的基因表达信息和第一预测模型输入的基因表达信息之间的差异,以及重构的空间信息与第一预测模型的输入中的空间信息之间的差异,基于该损失函数对第一预测模型进行训练,能够更准确且全面地迭代训练第一预测模型中的模型参数,使得得到的调整后的第一预测模型更优,进而使得确定的细胞注释结果准确度更高。
[0132]
在本技术的另一实施例中,在模型训练过程中,还可以为损失函数中第一分量、第二分量以及第三分量分配合理的权重系数,使得模型的预测差异与实际业务需求高度匹配,也能带来模型性能的提升。在一种可能的实现方式中,在确定损失函数时,可以通过确定第一分量、第二分量以及第三分量的权重系数,根据第一分量、第二分量、第三分量的权重系数、第一分量、第二分量以及第三分量确定损失函数。其中,上述第一分量的权重系数与转录组细胞注释结果的重要程度相关;第二分量的权重系数与基因表达信息的重要程度相关;第二分量的权重系数与转录组空间信息的重要程度相关。
[0133]
其中,第一分量的权重系数与转录组细胞注释结果的重要程度呈正相关,即第一分量的权重系数越大,则表示转录组细胞注释结果的重要程度越高。同理,第二分量的权重系数与转录组基因表达信息的重要程度呈正相关,即第二分量的权重系数越大,则表示转录组基因表达信息的重要程度越高。第三分量的权重系数与转录组空间信息的重要程度呈正相关,即第三分量的权重系数越大,则表示转录组空间信息的重要程度越高。
[0134]
上述第一分量、第二分量、第三分量以及损失函数之间满足以下公式:
[0135]
y=a1*y1+a2*y2+a3*y3[0136]
其中,y为上述第一预测模型训练过程中的损失函数,y1为第一分量,a1为第一分量的权重系数,y2为第二分量,a2为第二分量的权重系数,y3为第三分量,a3为第三分量的权重系数。,
[0137]
本技术实施例中,还可以根据细胞数据注释方法应用的业务场景,合理确定第一分量、第二分量和第三分量的权重系数。一种可能的实现方式中,根据业务需求确定转录组空间信息、转录组基因表达信息和转录组细胞注释结果的重要程度比例,并根据重要程度比例确定第一分量、第二分量和第三分量的权重系数。
[0138]
示例性地,将本技术实施例提供的细胞数据注释方法应用于某些场景中,更为关注转录组细胞注释结果,例如,转录组细胞注释结果的重要程度、转录组基因表达信息的重要程度和转录组空间信息的重要程度比例为80%:10%:10%,则第一分量的权重系数可以是0.8,第二分量的权重系数可以是0.1,第三分量的权重系数可以是0.1。
[0139]
在本技术的另一实施例中,还提供了根据待预测转录组的空间信息确定邻接矩阵的具体实现方式。可以根据空间信息确定多个测序点中每一测序点的邻近测序点,然后根据邻近测序点与对应测序点之间的空间距离生成邻接矩阵,该邻接矩阵为n*n矩阵,其中n
为待预测转录组中测序点的数量。
[0140]
具体地,上述空间信息用于表示每个测序点在切片中的位置信息,该空间信息可以通过矩阵表示,例如可以是二维矩阵,也可以是三维矩阵。当空间信息为二维矩阵时,可以是n*2维的坐标矩阵,其中,矩阵中每行的元素值表示每个测序点的横坐标值和纵坐标值,每列的元素值表示多个测序点。当空间信息为三维矩阵时,可以是n*3维的坐标矩阵。需要说明的是,上述邻近测序点的数量可以是多个。
[0141]
其中,根据空间信息确定多个测序点中每一测序点的邻近测序点时,可以先基于空间信息构建对应的空矩阵,例如空间信息为n*2维的坐标矩阵,则构建的空矩阵为n*n维的坐标矩阵,其中,n为测序点数量。然后在空矩阵中确定每一测序点的邻域范围,将邻域范围内的测序点确定为该测序点的邻近测序点,可以是从空矩阵自上向下的顺序,先从第一个测序点开始,确定第一个的测序点对应的邻近测序点,然后再确定第二个的测序点对应的邻近测序点,以此类推,确定最后一个的测序点对应的邻近测序点,从而确定出每个测序点对应的邻近测序点。该邻域范围可以是根据该测序点的位置信息按照预设规则确定的,例如是以测序点的位置信息为中心,以预设距离为半径的区域。
[0142]
作为一种可实现方式,根据空间信息确定多个测序点中每一测序点的邻近测序点时,可以是针对多个测序点中的每一测序点,根据空间信息确定测序点的位置信息,并根据测序点的位置信息计算测序点与其余测序点之间的距离,然后将与测序点之间距离小于预设阈值的测序点,确定为测序点的邻近测序点。其中,该预设阈值可以是根据实际需求自定义设置的。
[0143]
进一步地,对于多个测序点中的每一测序点,在确定出对应的邻近测序点后,可以确定邻近测序点与对应测序点之间的空间距离,例如可以通过调用距离计算函数得到空间距离。然后根据邻近测序点与对应测序点之间的空间距离,对空间距离进行归一化处理,得到该测序点与其他测序点的元素,该元素用于表示该测序点与其他测序点的邻近关系,从而构建邻接矩阵。
[0144]
具体地,可以是从空矩阵自上向下的顺序,先从第一个测序点开始,确定第一个的测序点与对应的邻近点之间的空间距离,然后对空间距离进行归一化处理,得到处理结果,并将该处理结果作为该第一个测序点与其邻近测序点在空矩阵中对应位置处的元素,然后确定第二个的测序点与对应的邻近点之间的空间距离,并进行归一化处理,得到第二个测序点与其邻近测序点在空矩阵中对应位置处的元素,以此类推,确定最后一个测序点与其邻近测序点在空矩阵中对应位置处的元素,从而得到邻接矩阵。
[0145]
作为一种可实现方式,针对多个测序点中的每一测序点,可以根据测序点的邻近测序点对应的空间距离,确定邻接矩阵中与测序点对应的元素,通过根据多个测序点中第i个测序点对应的k个邻近测序点确定邻接矩阵中第i行或第i列的k个元素。然后将邻接矩阵中的其余元素值为零。
[0146]
具体地,在确定出每一测序点对应的邻近测序点后,对于每一测序点,可以确定邻近测序点的位置信息,根据该测序点与邻近测序点的位置信息计算两者之间的空间距离。其中,当空间信息为二维矩阵时,构建的空矩阵为n*n维矩阵时,则任一测序点的位置信息可以通过横坐标值和纵坐标值表示。在确定邻近测序点与对应测序点之间的空间距离时,可以是通过计算该测序点与各邻近测序点之间的欧式距离,从而将该欧式距离作为空间距
离。然后对欧式距离取相反数并归一化处理至[0,1]的范围内的值,将该值作为邻接矩阵中与测序点对应的元素,并对于邻接矩阵中的其余元素置为零。
[0147]
示例性地,当空间信息为n*2维矩阵时,可以建立一个n*n维的空矩阵,其中,n代表测序点个数,即细胞数量,该矩阵中的每一行代表一个测序点或细胞,并对于多个测序点中的每一测序点i,根据空间信息确定测序点i的位置信息,该位置信息可以包括横坐标值和纵坐标值,然后根据测序点i的位置信息计算测序点i与其余测序点之间的距离,并将与测序点i之间距离小于预设阈值的测序点,确定为测序点i的邻近测序点,例如确定出k个测序点j为邻近测序点,包括j1,j2,
…
,jk,然后对于多个邻近测序点中的每个邻近测序点,计算测序点i与邻近测序点j之间的欧式距离,并计算欧式距离的相反数并归一化处理为[0,1]的范围内的值,从而得到空矩阵在(i,j)位置对应的元素值,而对于不在邻域内的点,即非邻近测序点在邻接矩阵中对应位置的元素值置为零,从而生成距离加权邻接矩阵,将该邻接矩阵记为s,为n阶矩阵。需要说明的是,该邻接矩阵中所有元素均在[0,1]的范围内,邻接矩阵的每一行代表每一个测序点或细胞的邻域关系,每行中均有k个元素不为零,为零代表该位置的细胞或测序点与本行所代表的细胞或测序点不相邻,而元素在不为零时,值越大代表该位置的细胞或测序点与本行的细胞或测序点距离越近。
[0148]
为了更好的理解本技术实施例,下面来进一步说明本技术提出的细胞数据注释的方法的完整流程图方法。
[0149]
如图9所示,该方法可以包括以下步骤:
[0150]
s401、获取待预测转录组的细胞数据;细胞数据包括待预测转录组中多个测序点的基因表达信息以及多个测序点的空间信息。
[0151]
具体地,请参见图10所示,上述待预测转录组的细胞数据(st dataset)包括多个测序点的基因表达信息和多个测序点的空间信息,其中,每个测序点的基因表达信息可以表示为n
×
g维的矩阵,其中,n为测序点个数,g为测到的基因数,矩阵中的第i行第j列表示在测序点i上基因j的表达量。上述多个测序点中每个测序点的空间信息是指每个测序点在切片中的坐标信息,该坐标信息可以为二维数据,也可以为三维数据。以二维数据为例,该坐标信息可以为n
×
2维的矩阵,其中,n为测序点个数,其中,矩阵中的第i行表示测序点i在切片中的位置坐标,可以包括两个横坐标值和纵坐标值。其中,可以将待预测转录组的基因表达信息对应的矩阵记为x。
[0152]
s402、确定与待预测转录组对应的已注释细胞对象以及与细胞对象匹配的第二预测模型。
[0153]
s403、将基因表达信息输入第二预测模型,获得待预测转录组的初始细胞注释结果。
[0154]
具体的,待预测转录组对应的细胞对象是指组织或区域与待预测转录组相同的细胞对象,例如,待预测转录组为取自脑部组织的大脑切片,则与该待预测转录组对应细胞对象也取自脑部组织。上述细胞对象可以是单细胞或细胞组。
[0155]
以该待预测转录组对应的细胞对象为单细胞数据,与单细胞匹配的第二预测模型为dnn模型为例,该单细胞数据(single-cell dataset)可以包括两部分,第一部分是单细胞的基因表达信息,为m*n维矩阵,其中,n为测序点或细胞个数,g为测到的基因数,第二部分为每个单细胞对应的细胞注释结果,即细胞类型,为m维向量。其中,可以将单细胞的基因
表达信息对应的矩阵记为a,其对应的细胞类型记为y。
[0156]
其中,在单细胞迁移阶段,可以通过单细胞数据集和对应的细胞注释结果进行训练(training)得到dnn模型,通过将单细胞数据集按照一定比例随机分为训练集和验证集,然后利用训练集和验证集按照训练学习算法构建得到dnn模型。
[0157]
具体地,在构建得到dnn模型后,将待预测转录组的细胞数据(single-cell dataset)输入该dnn模型中进行预测(inference)处理,可以是通过提取到待预测转录组的特征信息,然后对特征信息进行运算,从而得到待预测转录组的初始细胞注释结果,即细胞类型,该细胞类型可以通过待预测转录组中每个细胞类型概率(class probabilities for each cells of st dataset)表示,该初始细胞注释结果可以通过n*c维的矩阵表示,其中,n为待预测转录组的测序点或细胞数量,c为细胞类型数量,可以将该初始细胞注释结果记为l。
[0158]
可以理解的是,上述n*c维的矩阵中的每一行对应一个测序点或细胞,每一列对应一个细胞类型。若某一行某一列的值较大,则表示该行对应的细胞有可能属于该列对应的细胞类型。其中,该初始细胞注释结果l可以作为第一预测模型的伪标签。
[0159]
s404、对细胞数据输入第一预测模型进行编码处理得到融合编码信息,将融合编码信息输入第一预测模型的分类模块,获得第一预测模型的输出结果。
[0160]
s405、根据第一预测模型的损失函数确定输出结果和初始细胞注释结果之间的损失,根据损失迭代训练分类模块,在损失函数最小化时获得调整后的第一预测模型。
[0161]
需要说明的是,上述单细胞的基因表达信息对应的矩阵a和对应的细胞类型y,以及待预测转录组的基因表达信息对应的矩阵x这三个部分可以直接被相应的第一预测模型使用。但是待预测转录组的空间信息,即n*2维的坐标矩阵需要转换为对应的距离加权邻接矩阵,从而将该邻接矩阵作为第一预测模型输入的一部分。
[0162]
本实施例中,上述待预测转录组的空间信息为n*2维的坐标矩阵,可以先建立n*n维的空矩阵,其中,空矩阵的每一行代表一个测序点,然后对于每个测序点i取其预设距离范围内的k个测序点j1,j2,
…
,jk作为该测序点i的邻近测序点,然后对于多个邻近测序点中的每个邻近测序点,计算测序点i与邻近测序点j之间的欧式距离,并计算欧式距离的相反数并归一化处理为[0,1]的范围内的值,从而得到空矩阵在(i,j)位置对应的元素值,而对于不在邻域内的点,即非邻近测序点在邻接矩阵中对应位置的元素值置为零,从而生成距离加权邻接矩阵,将该邻接矩阵记为s,为n阶矩阵。
[0163]
请参见图11所示,该第一预测模型可以包括两对编码-解码模块和一个分类模块组成,其中,一个编码-解码模块可以是dae,用于获取对基因表达信息进行特征编码,得到第一编码信息,该dae包括deep encoder和deep decoder;另一个编码-解码模块可以是vgae,用于获取对空间信息进行特征编码,得到第二编码信息,该vgae包括graph encoder和graph decoder;上述分类模块用于负责建立完整的特征编码与目标细胞类型之间的对应关系,可以包括cluster/classifier。
[0164]
将待预测转录组的基因表达信息对应的矩阵x、邻接矩阵s输入第一预测模型中两个编码-解码模块和一个分类模块中,待预测转录组的基因表达信息对应的矩阵x通过dae的第一编码模块(deep encoder)进行编码处理,得到第一编码信息e
x
,将邻接矩阵s和第一编码信息e
x
通过vgae中的第二编码模块(graph encoder)进行编码处理,得到第二编码信
息eg,然后将第一编码信息e
x
和第二编码信息eg进行融合编码处理,可以是通过拼接处理得到融合编码信息e。可以理解的是,上述融合编码信息e同时包含了待预测转录组中的基因表达信息对应的矩阵x和空间信息对应的邻接矩阵s编码的信息。
[0165]
需要说明的是,对第一预测模型进行训练的最终目标是为了使得融合特征编码e能够有效编码待预测转录组中的所有信息,并能够被分类模块准确得分类处理得到对应的细胞类型。为此,引入对应的两个解码模块通过自监督的形式提高融合特征编码e的鲁棒性。其中,自监督是指利用自身的信息作为标签,通过编码-解码的形式以最大程度地使得解码后得到的结果与编码前输入的结果一致。
[0166]
进一步地,将融合编码信息e可以经过两个线性层进行特征提取处理后得到第一中间特征信息和第二中间特征信息,然后将第一中间特征信息通过dae的第一解码模块(deep decoder)进行特征还原处理,得到重构的基因表达信息x’,该重构的基因表达信息通过n*g维的矩阵表示,n为测序点或细胞个数,g为测到的基因数,并行地将第二中间特征信息通过vgae的第二解码模块(graph decoder)进行特征还原处理,得到重构的空间信息s’,该重构的空间信息通过n*n维的矩阵表示,n为测序点或细胞个数,并将融合编码信息e通过分类模块(cluster/classifier)进行分类处理,生成第一预测模型的输出结果l’,该输出结果可以通过n*c维的矩阵表示,n为待预测转录组的测序点或细胞数量,c为细胞类型数量。
[0167]
进一步地,根据第一预测模型的损失函数确定输出结果与初始细胞注释结果l之间的损失,根据损失迭代训练第一预测模型,在损失函数最小化时获得调整后的第一预测模型。上述损失函数包括第一分量、第二分量和第三分量。第一分量为初始细胞注释结果l与第一预测模型的输出l’之间的分类损失,第二分量为重构的基因表达信息x’与第一预测模型中输入中的基因表达信息x之间的重构损失,第三分量为重构空间信息s’与第一预测模型的输入中的空间信息对应的邻接矩阵s之间的重构损失。可选的,在模型训练过程中,还可以为损失函数中的第一分量、第二分量以及第三分量分配合理的权重系数,可以为确定第一分量的权重系数是0.8,第二分量的权重系数可以是0.1,第三分量的权重系数可以是0.1,则可以根据第一分量与第一分量的权重系数、第二分量与第二分量的权重系数、第三分量和第三分量的权重系数确定损失函数。然后按照损失函数最小化进行迭代训练第一预测模型,可以是采用梯度下降法对第一预测模型中的第一编码模块、第二编码模块和分类模块中的参数进行调整,从而得到调整后的第一预测模型。
[0168]
本实施例中通过设置第一分量、第二分量和第三分量构建损失函数,能够通过优化三部分损失函数,使得准确地对第一预测模型中的模型参数进行调整,从而使得调整后的第一预测模型达到更优,进而能够实现对待预测转录组的细胞数据进行更准确地注释。
[0169]
s406、将细胞数据输入调整后的第一预测模型,获得待预测转录组的细胞注释结果。
[0170]
具体地,请参见图12所示,上述调整后第一预测模型包括调整后的编码模块和分类模块cls。其中,该调整后的编码模块包括第三编码模块(d-enc)和第四编码模块(g-enc)。在得到调整后的第一预测模型后,可以将细胞数据st dataset输入调整后的第一预测模型,将基因表达信息x输入调整后的第一预测模型中的第三编码模块(d-enc),获得第三编码信息,然后根据空间信息确定邻接矩阵s,该邻接矩阵s为n阶矩阵,并将邻接矩阵s和
第三编码信息输入调整后的第一预测模型的第四编码模块(g-enc),获得第四编码信息,将第三编码信息和第四编码信息进行融合处理,得到合并编码信息。将合并编码信息输入调整后的第一预测模型的分类模块(cls)进行分类处理,得到待预测转录组的细胞注释结果,该细胞注释结果可以包括细胞类型(cell type annotation)。该细胞注释结果可以通过n*c维的矩阵表示,n为待预测转录组的测序点或细胞数,c为细胞类型数。
[0171]
另外,为了对本技术中方案的性能进行测试,通过分别计算出seurat、scmap、scnym、scibet等不同方案对merfish和slide-seq两个空间转录组数据的准确率指标,其中,seurat、scmap、scnym、scibet表示现有技术中得到的方案,seurat和scmap表示基于参考数据集的注释方案,该方案是指先对待预测数据集进行聚类处理,得到聚类结果(即被分为多个簇),然后根据参考数据集确定每个簇对应的细胞注释结果;scnym、scibet表示基于分类训练的方法,该准确度指标包括得到如下数据:
[0172] 本方案seuratscmapscnymscibetmerfish92.21%86.80%72.52%87.72%90.43%slide-seq60.76%57.76%20.71%52.69%47.87%
[0173]
通过以上数据表示:本方案对于空间转录组的细胞数据进行注释的性能指标最好,即通过整合空间转录组中的基因表达信息和空间信息,从而更为精准、全面地利用空间转录组的细胞数数据中的所有信息,从而提高了细胞注释结果确定的准确性。
[0174]
本实施例中能够有效地融合了多个测序点的基因表达信息以及多个测序点的空间信息,结合指导信息进行自监督修正,从而更为精准、全面地利用待预测转录组的细胞数据中的所有信息,进而更加准确地获得待预测转录组的细胞注释结果,很大程度上提升了对待预测转录组的细胞数据注释的准确度。
[0175]
另一方面,图13为本技术实施例提供的一种细胞数据注释装置的结构示意图。该装置可以为终端或服务器内的装置,如图13所示,该装置700包括:
[0176]
获取模块710,用于获取待预测转录组的细胞数据;细胞数据包括待预测转录组中多个测序点的基因表达信息以及多个测序点的空间信息;
[0177]
处理模块720,用于确定与待预测转录组对应的已注释细胞对象,根据细胞对象以及基因表达信息确定待预测转录组的初始细胞注释结果;
[0178]
细胞注释模块730,将细胞数据输入第一预测模型,根据第一预测模型的输出与初始细胞注释结果之间的损失对初始细胞注释结果进行修正,获得待预测转录组的细胞注释结果。
[0179]
在一些实施例中,请参见图14所示,上述处理模块720,包括:
[0180]
第一确定单元721,用于确定与细胞对象匹配的第二预测模型;第二预测模型是基于细胞对象的历史基因表达信息以及历史基因表达信息对应的历史细胞注释结果进行训练获得的;
[0181]
第一处理单元722,用于将基因表达信息输入第二预测模型,获得待预测转录组的初始细胞注释结果。
[0182]
在一些实施例中,细胞对象为待预测转录组对应的基因测序对象包括的单细胞或细胞组。
[0183]
在一些实施例中,上述细胞注释模块730,包括:
[0184]
第二处理单元731,用于对细胞数据输入第一预测模型进行编码处理得到融合编码信息,将融合编码信息输入第一预测模型的分类模块,获得第一预测模型的输出结果;
[0185]
训练单元732,用于根据第一预测模型的损失函数确定输出结果和初始细胞注释结果之间的损失,根据损失迭代训练分类模块,在损失函数最小化时获得调整后的第一预测模型;
[0186]
第二确定单元733,用于将细胞数据输入调整后的第一预测模型,获得待预测转录组的细胞注释结果。
[0187]
在一些实施例中,上述第二处理单元731,具体用于:
[0188]
将基因表达信息输入第一预测模型的第一编码模块,获得第一编码信息;
[0189]
根据空间信息确定邻接矩阵;邻接矩阵用于表征待预测转录组中各个测序点的邻近测序点;
[0190]
将邻接矩阵和第一编码信息输入第一预测模型的第二编码模块进行处理,获得第二编码信息;
[0191]
将第一编码信息和第二编码信息进行融合处理,得到融合编码信息。
[0192]
在一些实施例中,上述第二处理单元731,还用于:
[0193]
对融合编码信息进行解码处理,获得重构的基因表达信息;
[0194]
根据损失函数确定重构的基因表达信息与基因表达信息之间的损失,根据损失调整第一编码模块的参数,使得重构的基因表达信息接近基因表达信息。
[0195]
在一些实施例中,上述第二处理单元731,还用于:
[0196]
对融合编码信息进行线性特征提取处理,得到中间特征信息;
[0197]
将中间特征信息输入通过第一解码器进行特征还原处理,获取重构的基因表达信息。
[0198]
在一些实施例中,上述第二处理单元731,还用于:
[0199]
对融合编码信息进行解码处理,获得重构的空间信息;
[0200]
根据损失函数确定重构的空间信息与空间信息之间的损失,根据损失调整第二编码模块的参数,使得重构的空间信息接近空间信息。
[0201]
在一些实施例中,损失函数包括第一分量、第二分量以及第三分量;
[0202]
第一分量用于表征初始细胞注释结果与第一预测模型的输出之间的损失;
[0203]
第二分量用于表征重构的基因表达信息与第一预测模型的输入中的基因表达信息之间的损失;重构的基因表达信息是对融合编码信息进行解码处理后获得的;
[0204]
第三分量用于表征重构的空间信息与第一预测模型的输入中的空间信息之间的损失;重构的空间信息是对融合编码信息进行解码处理后获得的。
[0205]
在一些实施例中,上述装置,还用于:
[0206]
确定第一分量、第二分量以及第三分量的权重系数;
[0207]
根据第一分量、第二分量、第三分量的权重系数、第一分量、第二分量以及第三分量确定损失函数;
[0208]
其中,第一分量的权重系数与转录组细胞注释结果的重要程度相关;第二分量的权重系数与转录组基因表达信息的重要程度相关;第三分量的权重系数与转录组空间信息的重要程度相关。
[0209]
在一些实施例中,上述第二处理单元731,还用于:
[0210]
根据空间信息确定多个测序点中每一测序点的邻近测序点;
[0211]
根据待预测转录组中所有测序点的邻近测序点所对应空间距离生成邻接矩阵;邻接矩阵为n*n矩阵,n为待预测转录组中测序点的数量。
[0212]
在一些实施例中,上述第二处理单元731,还用于:
[0213]
针对多个测序点中的每一测序点,根据空间信息确定测序点的位置信息,根据测序点的位置信息计算测序点与待预测转录组中其余测序点之间的距离;
[0214]
将与测序点之间距离小于预设阈值的测序点,确定为测序点的邻近测序点。
[0215]
在一些实施例中,上述第二处理单元731,还用于:
[0216]
针对多个测序点中的每一测序点,根据测序点与邻近测序点对应的空间距离,确定邻接矩阵中与测序点对应的元素;
[0217]
将邻接矩阵中的其余元素置为零。
[0218]
可以理解的是,本实施例的细胞数据注释装置的各功能模块的功能可根据上述方法实施例中的方法具体实现,其具体实现过程可以参照上述方法实施例的相关描述,在此不再赘述。
[0219]
综上所述,本技术实施例提供的细胞数据注释装置,通过确定与待预测转录组对应的已注释细胞对象,并根据细胞对象以及基因表达信息确定待预测转录组的初始细胞注释结果,其在无需任何人工参与注释的情况下,能够获取到待预测转录组的细胞注释结果的指导信息,并且通过第一预测模型对细胞数据进行预测,根据第一预测模型的输出与初始细胞注释结果之间的损失对初始细胞注释结果进行修正,有效地融合了多个测序点的基因表达信息以及多个测序点的空间信息,能够结合指导信息进行自监督修正,从而更为精准、全面地利用待预测转录组的细胞数据中的所有信息,进而更加准确地获得待预测转录组的细胞注释结果,很大程度上提升了对待预测转录组的细胞数据注释的准确度。还可以应用于测序分析系统中,对待预测转录组的细胞数据进行准确的预测,极大地提升了细胞注释的质量和效率,为空间转录组的数据分析提供了强有力的支持。
[0220]
另一方面,本技术实施例提供的设备,包括存储器、处理器以及存储在存储器上并可在处理器上运行的计算机程序,该处理器执行该程序时实现如上述的细胞数据注释方法。
[0221]
下面参考图15,图15为本技术实施例的终端设备的计算机系统的结构示意图。
[0222]
如图15所示,计算机系统300包括中央处理单元(cpu)301,其可以根据存储在只读存储器(rom)302中的程序或者从存储部分303加载到随机访问存储器(ram)303中的程序而执行各种适当的动作和处理。在ram 303中,还存储有系统300操作所需的各种程序和数据。cpu 301、rom 302以及ram 303通过总线304彼此相连。输入/输出(i/o)接口305也连接至总线304。
[0223]
以下部件连接至i/o接口305:包括键盘、鼠标等的输入部分306;包括诸如阴极射线管(crt)、液晶显示器(lcd)等以及扬声器等的输出部分307;包括硬盘等的存储部分308;以及包括诸如lan卡、调制解调器等的网络接口卡的通信部分309。通信部分309经由诸如因特网的网络执行通信处理。驱动器310也根据需要连接至i/o接口305。可拆卸介质311,诸如磁盘、光盘、磁光盘、半导体存储器等等,根据需要安装在驱动器310上,以便于从其上读出
的计算机程序根据需要被安装入存储部分308。
[0224]
特别地,根据本技术的实施例,上文参考流程图描述的过程可以被实现为计算机软件程序。例如,本技术的实施例包括一种计算机程序产品,其包括承载在机器可读介质上的计算机程序,该计算机程序包含用于执行流程图所示的方法的程序代码。在这样的实施例中,该计算机程序可以通过通信部分303从网络上被下载和安装,和/或从可拆卸介质311被安装。在该计算机程序被中央处理单元(cpu)301执行时,执行本技术的系统中限定的上述功能。
[0225]
需要说明的是,本技术所示的计算机可读介质可以是计算机可读信号介质或者计算机可读存储介质或者是上述两者的任意组合。计算机可读存储介质例如可以是——但不限于——电、磁、光、电磁、红外线、或半导体的系统、装置或器件,或者任意以上的组合。计算机可读存储介质的更具体的例子可以包括但不限于:具有一个或多个导线的电连接、便携式计算机磁盘、硬盘、随机访问存储器(ram)、只读存储器(rom)、可擦式可编程只读存储器(eprom或闪存)、光纤、便携式紧凑磁盘只读存储器(cd-rom)、光存储器件、磁存储器件、或者上述的任意合适的组合。在本技术中,计算机可读存储介质可以是任何包含或存储程序的有形介质,该程序可以被指令执行系统、装置或者器件使用或者与其结合使用。而在本技术中,计算机可读的信号介质可以包括在基带中或者作为载波一部分传播的数据信号,其中承载了计算机可读的程序代码。这种传播的数据信号可以采用多种形式,包括但不限于电磁信号、光信号或上述的任意合适的组合。计算机可读的信号介质还可以是计算机可读存储介质以外的任何计算机可读介质,该计算机可读介质可以发送、传播或者传输用于由指令执行系统、装置或者器件使用或者与其结合使用的程序。计算机可读介质上包含的程序代码可以用任何适当的介质传输,包括但不限于:无线、电线、光缆、rf等等,或者上述的任意合适的组合。
[0226]
附图中的流程图和框图,图示了按照本技术各种实施例的系统、方法和计算机程序产品的可能实现的体系架构、功能和操作。在这点上,流程图或框图中的每个方框可以代表一个模块、程序段、或代码的一部分,前述模块、程序段、或代码的一部分包含一个或多个用于实现规定的逻辑功能的可执行指令。也应当注意,在有些作为替换的实现中,方框中所标注的功能也可以以不同于附图中所标注的顺序发生。例如,两个接连地表示的方框实际上可以基本并行地执行,它们有时也可以按相反的顺序执行,这依所涉及的功能而定。也要注意的是,框图和/或流程图中的每个方框、以及框图和/或流程图中的方框的组合,可以用执行规定的功能或操作的专用的基于硬件的系统来实现,或者可以用专用硬件与计算机指令的组合来实现。
[0227]
描述于本技术实施例中所涉及到的单元或模块可以通过软件的方式实现,也可以通过硬件的方式来实现。所描述的单元或模块也可以设置在处理器中,例如,可以描述为:一种处理器,包括:获取模块、处理模块及细胞注释模块。其中,这些单元或模块的名称在某种情况下并不构成对该单元或模块本身的限定,例如,获取模块还可以被描述为“用于获取待预测转录组的细胞数据;所述细胞数据包括所述待预测转录组中多个测序点的基因表达信息以及所述多个测序点的空间信息”。
[0228]
作为另一方面,本技术还提供了一种计算机可读存储介质,该计算机可读存储介质可以是上述实施例中描述的电子设备中所包含的;也可以是单独存在,而未装配入该电
子设备中的。上述计算机可读存储介质存储有一个或者多个程序,当上述前述程序被一个或者一个以上的处理器用来执行描述于本技术的细胞数据注释方法:
[0229]
获取待预测转录组的细胞数据;所述细胞数据包括所述待预测转录组中多个测序点的基因表达信息以及所述多个测序点的空间信息;
[0230]
确定与所述待预测转录组对应的已注释细胞对象,根据所述细胞对象以及所述基因表达信息确定所述待预测转录组的初始细胞注释结果;
[0231]
将所述细胞数据输入第一预测模型,根据所述第一预测模型的输出与所述初始细胞注释结果之间的损失对所述初始细胞注释结果进行修正,获得所述待预测转录组的细胞注释结果。
[0232]
综上所述,本技术实施例中提供的细胞数据注释方法、装置、设备及介质,通过确定与待预测转录组对应的已注释细胞对象,并根据细胞对象以及基因表达信息确定待预测转录组的初始细胞注释结果,其在无需任何人工参与注释的情况下,能够获取到待预测转录组的细胞注释结果的指导信息,并且通过第一预测模型对细胞数据进行预测,根据第一预测模型的输出与初始细胞注释结果之间的损失对初始细胞注释结果进行修正,有效地融合了多个测序点的基因表达信息以及多个测序点的空间信息,能够结合指导信息进行自监督修正,从而更为精准、全面地利用待预测转录组的细胞数据中的所有信息,进而更加准确地获得待预测转录组的细胞注释结果,很大程度上提升了对待预测转录组的细胞数据注释的准确度。还可以应用于测序分析系统中,对待预测转录组的细胞数据进行准确的预测,极大地提升了细胞注释的质量和效率,为空间转录组的数据分析提供了强有力的支持。
[0233]
以上描述仅为本技术的较佳实施例以及对所运用技术原理的说明。本领域技术人员应当理解,本技术中所涉及的发明范围,并不限于上述技术特征的特定组合而成的技术方案,同时也应涵盖在不脱离所述发明构思的情况下,由上述技术特征或其等同特征进行任意组合而形成的其它技术方案。例如上述特征与本技术中公开的(但不限于)具有类似功能的技术特征进行互相替换而形成的技术方案。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1