基于stacking集成学习策略的Cas蛋白预测方法
基于stacking集成学习策略的cas蛋白预测方法
技术领域
1.本发明属于生物信息学领域,具体涉及一种cas蛋白预测方法。
背景技术:
2.crispr-cas系统是大多数细菌与古菌中用于抵御病毒的一种免疫系统。在不断探究crispr-cas系统及其功能多样性的相关研究中最关键的步骤之一是鉴定crispr-cas系统中的cas蛋白。在过去的10年里,随着科学家们对crispr-cas系统的深入理解,目前已在细菌和古细菌基因组中鉴定得到了与crispr-cas系统相关的不同蛋白家族至少45个。由rna引导的dna内切酶,如cas9和c2c2等cas蛋白,越来越多的被应用于基因组编辑领域。除此之外,基于crispr进行病毒诊断的相关研究也不断兴起,但与基因编辑领域常用cas9不同的是,基于crispr的诊断研究中常用cas13这一家族。有效识别更多的cas蛋白不仅是帮助生物学家进一步推断crispr-cas系统类型,理解crispr-cas系统机制的必要条件,也是支持基因编辑新型剪子和基于crispr的病毒诊断研究等新型技术发展的起点。目前为止,已经开发了一些基于机器学习的方法来识别cas蛋白,但其在模型构建及特征工程方面存在着一定的局限性,导致无法达到理想的预测效果。因此,如何表征cas蛋白与非cas蛋白之间的分类特征对于准确识别cas蛋白变得极其重要。当前急需利用计算方法找到用于鉴别cas蛋白的关键特征并据此开发出更有效的cas蛋白预测模型来帮助研究人员从巨量的蛋白质组学数据中找到更多假定的cas蛋白,提高识别效率,为crispr-cas系统的研究提供更多的数据资源。
技术实现要素:
3.本发明是为了解决目前cas蛋白识别方法中在模型构建及特征工程方面存在着一定的局限性,导致无法达到理想的预测效果问题。
4.基于stacking集成学习策略的cas蛋白预测方法,利用双层预测模型对潜在cas蛋白进行识别;所述双层预测模型通过以下步骤确定:
5.s1、获取cas蛋白序列数据集;
6.s2、使用基于序列模式特征、进化信息特征、理化性质特征、深度表示学习特征的编码对cas蛋白数据进行编码,构建初始特征空间;
7.基于序列模式特征包括aac、qso、asdc、cksaap、dde、dpc;进化信息特征包括pse-pssm、tri-gram-pssm、aatp;理化性质特征包括ctdc、ctdt;深度表示学习特征包括unirep;
8.aac为氨基酸组成,通过计算每一种氨基酸残基出现的频率编码序列;
9.qso为准序列描述符,通过氨基酸间的物理化学距离表征蛋白质序列的顺序信息;
10.asdc为自适应跳跃二肽组成,是一个400维的向量,每一维表示为氨基酸序列长度内所有可能残基对的出现频率;
11.cksapp为k间隔氨基酸对组成,即有k个氨基酸残基间隔的氨基酸对在序列中出现的频率;
12.dde为二肽与预期平均值的偏差,通过计算二肽频率与期望平均值偏差生成的400维特征向量;
13.dpc为二肽组成,通过对蛋白质序列所有可能的二肽的分布情况进行编码,使用氨基酸序列的片段信息来表征其特征信息;
14.pse-pssm是反映序列中一个氨基酸和它后面的α个氨基酸之间的关系向量,用表示,其通过以下方式计算得到:
[0015][0016][0017][0018][0019]
其中,l表示蛋白质序列的长度,e
i,j
表示pssm第i行第j列中的元素;t
i,j
、为中间参量;
[0020]
tri-gram-pssm是通过蛋白质序列的pssm profile中氨基酸的替换概率来计算每个tri-gram的特征向量,向量中的每一维通过以下公式计算得到:
[0021][0022]
其中,p
i,j
表示pssm中第i行、第j列的元素值,l为蛋白质序列的长度;m、n、r分别表示与i、i+1,i+2的氨基酸各自相邻一定距离的氨基酸,且取值都是1-20;
[0023]
aatp为aac-pssm生成的维向量与转移概率组合生成的向量组合得到的向量;aac-pssm生成的维向量是通过位置特异性评分矩阵进行aac-pssm特征编码得到的;转移概率组合生成的向量是通过将转移概率矩阵tpm导入pssm来计算相邻氨基酸残基的关系进行编码得到的;
[0024]
ctdc为按照物理化学性质归属于某类氨基酸的数量与氨基酸序列的长度比;
[0025]
ctdt为基于物理化学性质,不同类型残基之间相邻的频率;
[0026]
unirep为使用multiplicative long-/short-term-memory rnns得到的特征;
[0027]
s3、通过基于lgbm算法的三步特征空间优化策略对初始特征空间进行特征空间优化,得到最优特征空间;
[0028]
s4、利用最优特征空间构建多个基学习模型,然后将多个基学习模型输出的置信度得分特征作为第二层模型的输入特征,引入svm算法进行二次建模,构建用于预测cas蛋白的双层预测模型。
[0029]
进一步地,利用双层预测模型对潜在cas蛋白进行识别之后,利用hmmer算法对识别为cas蛋白的样本在hmm序列数据库中进行搜索,进行cas蛋白的功能域注释。
[0030]
进一步地,所述qso的获取过程如下:
[0031]
分别针对schneider-wrede物理化学距离矩阵和grantham化学距离矩阵,利用如下述公式进行计算以表征蛋白质序列的顺序信息:
[0032][0033]
其中,xr和xd为计算准序列描述符的特征量,r表示氨基酸的种类;fr表示氨基酸r的标准化出现次数;i+d表示与第i个氨基酸距离为d的氨基酸,dist
i,i+d
表示序列中第i个氨基酸与第(i+d)个氨基酸之间的距离,n表示序列中的氨基酸总数,ω表示加权系数。
[0034]
进一步地,asdc中每一维表示的氨基酸序列长度内所有可能残基对的出现频率如下:
[0035][0036]
其中,l表示蛋白质序列的长度,表示表示蛋白质序列中第t-gap二肽的出现次数。
[0037]
进一步地,组合得到的向量aatp对应的aac-pssm和转移概率通过如下公式获得:
[0038][0039][0040]
其中,xj表示由aac-pssm生成的特征,y
i,j
表示由tpc生成的特征;l表示蛋白质序列的长度,p
i,j
表示pssm中第i行,第j列个元素。
[0041]
进一步地,所述ctdc通过以下方式获取:
[0042]
将氨基酸根据疏水性、归一化范德华体积、极性、极化率、二级结构、可溶性、电荷共七种类型的物理化学性质分为三种类别,用classa,classb,classc分别表示;
[0043]
ctdc根据理化性质ca按照如下公式计算:
[0044][0045]
其中,na分别表示属于某类氨基酸的数量,n是氨基酸序列的长度。
[0046]
进一步地,ctdt通过以下方式获取:
[0047]
根据如下公式计算不同类型残基之间相邻的频率:
[0048][0049]
其中,n
ab
表示是属于classa的残基后紧随属于classb类的残基的出现频率,n
ba
表示属于classb类的残基后紧跟属于classa类的残基出现频率,n表示氨基酸序列的长度。
[0050]
进一步地,使用multiplicative long-/short-term-memory rnns得到的unirep的过程如下:
[0051]
对于一个含有l个氨基酸残基的样本,首先使用one-hot进行编码,并将其嵌入一个l
×
10的矩阵中;随后将矩阵作为multiplicative long-/short-term-memory rnns的输入得到unirep特征。
[0052]
进一步地,s3所述通过基于lgbm算法的三步特征空间优化策略对初始特征空间进行特征空间优化的过程包括以下步骤:
[0053]
首先通过python中lightgbm库中的feature_importances函数计算由基于序列模式特征、进化信息特征、理化性质特征、深度表示学习特征组成的cas蛋白初始特征集中每一维特征的特征重要性得分;
[0054]
然后将初始特征集按得分由高到低进行排序并选出初始特征集中得分高于平均得分的特征作为次优特征空间,即:将总特征集的每一维得分相加求平均得分,然后筛选出高于平均得分的特征集作为次优特征空间;随后应用顺序前向搜索方法在次优特征空间中按重要性得分由大到小向特征池中按维添加特征,并将能够获得最大马修斯相关系数的特征集作为最优特征空间。
[0055]
进一步地,s4中利用最优特征空间构建多个基学习模型过程中,所述多个基学习模型为lgbm算法、rf算法、ert算法、xgboost算法、gbdt算法对应的五个基学习模型。
[0056]
有益效果:
[0057]
(1)本发明首次将进化信息和深度表示学习的特征编码应用于cas蛋白的预测识别,并且实现了cas蛋白的准确识别,为基于crispr的生物技术的开发提供了先导支持。
[0058]
(2)本发明的基于stacking策略的集成学习模型,提高了cas蛋白预测的准确率。
[0059]
(3)本发明的模型泛化性能较好,可广泛应用于在原核生物蛋白质组数据中预测cas蛋白,具有较好的实用性能。
附图说明
[0060]
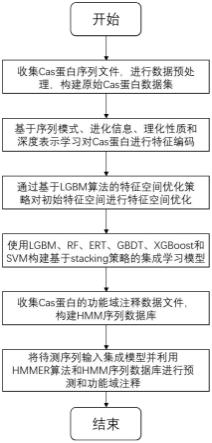
图1为实施例提供的基于stacking集成学习策略的cas蛋白预测方法流程图;
[0061]
图2(a)至图2(e)为实施例提供的训练数据集下xgboost、gbdt、rf、lgbm、ert分类模型对应的单一特征的分类效果示意图;
[0062]
图3(a)和图3(b)为实施例提供的特征空间优化前后的特征分布示意图;
[0063]
图4为实施例提供的训练数据集下的单一模型和集成模型的效果示意图;
[0064]
图5为实施例提供的与现有识别算法的识别效果对比示意图。
具体实施方式
[0065]
现在将参考附图来详细描述本发明的示例性实施方式。应当理解,附图中示出和描述的实施方式仅仅是示例性的,意在阐释本发明的原理和精神,而并非限制本发明的范围。
[0066]
具体实施方式一:结合图1说明本实施方式
[0067]
本实施方式所述的基于stacking集成学习策略的cas蛋白预测方法,包括以下步骤:
[0068]
s1、收集cas蛋白的序列数据文件,对数据进行去冗余等预处理和数据清洗过程,获取待处理的原始cas蛋白序列数据集;
[0069]
其中,获取的原始cas蛋白序列数据集包括正例数据集和反例数据集,正例数据集为cas蛋白序列文件,反例数据集为非cas蛋白序列文件。
[0070]
本发明实施例中,cas蛋白序列数据集由被分成了一个训练数据集(正例cas蛋白序列数量为150,反例cas蛋白序列数据为150)和一个测试数据集(正例cas蛋白序列数量为58,反例cas蛋白序列数据为58)。
[0071]
s2、使用基于序列模式特征、进化信息特征、理化性质特征、深度表示学习特征的十二种编码对cas蛋白数据进行编码,构建初始特征空间;
[0072]
基于序列模式特征包括aac、qso、asdc、cksaap、dde、dpc;
[0073]
进化信息特征包括pse-pssm、tri-gram-pssm、aatp;
[0074]
理化性质特征包括ctdc、ctdt;
[0075]
深度表示学习特征包括unirep;
[0076]
考虑到单一的特征提取方法很难准确识别cas蛋白,本发明实施例从序列模式、进化信息、理化性质和深度表征学习共四种视角中提取特征,综合评价模型的性能。所述特征如下:
[0077]
氨基酸组成(aac)通过计算每一种氨基酸残基出现的频率来编码序列;
[0078]
准序列描述符(qso)通过基于氨基酸间的物理化学距离表征蛋白质序列的顺序信息,其将下述公式分别应用于schneider-wrede物理化学距离矩阵和grantham化学距离矩阵进行计算:
[0079][0080]
其中,xr和xd为计算准序列描述符的特征量,r表示氨基酸的种类;fr表示氨基酸r的标准化出现次数;i+d表示与第i个氨基酸距离为d的氨基酸,dist
i,i+d
表示序列中第i个氨基酸与第(i+d)个氨基酸之间的距离,n表示序列中的氨基酸总数,ω表示加权系数,默认加权为0.1。
[0081]
自适应跳跃二肽组成(asdc)在考虑相邻氨基酸残基信息的同时,也考虑了残基之间的相关信息,最终产生一个400维的向量,其中每一维表示为氨基酸序列长度内所有可能
残基对的出现频率,其每一维的计算公式如下:
[0082][0083]
其中,l表示蛋白质序列的长度,表示表示蛋白质序列中第t-gap二肽的出现次数。
[0084]
k间隔氨基酸对的组成(cksapp)表示有k个氨基酸残基间隔的氨基酸对在序列中出现的频率,在本发明中采取的间隔为3;
[0085]
二肽与预期平均值的偏差(dde)描述符通过计算二肽频率与期望平均值的偏差,生成400维特征向量;
[0086]
二肽组成(dpc)通过对蛋白质序列所有可能的二肽的分布情况进行编码,使用氨基酸序列的片段信息来表征其特征信息。
[0087]
pse-pssm能够避免序列顺序信息的丢失,表征更有效的信息。它是一种可靠的特征编码方法,用于提取基于pssm变换的进化信息,它反映了序列中一个氨基酸和它后面的α个氨基酸之间的关系,其通过以下方式进行计算:
[0088][0089][0090][0091][0092]
其中,l表示蛋白质序列的长度,e
i,j
表示pssm第i行第j列中的元素,α使用默认值1。
[0093]
tri-gram-pssm通过蛋白质序列的pssm profile中氨基酸的替换概率来计算每个tri-gram来避免传统基于序列进行tri-gram特征编码会产生大多数零的问题,最终产生了一个大小为8000维的特征向量,其计算公式如下:
[0094][0095]
其中,p
i,j
表示pssm中第i行、第j列的元素值,l为蛋白质序列的长度。m、n、r表示三个相邻的氨基酸,且取值都是1-20,每一维特征的计算都要按照这个公式进行一遍累加。比如当m、n、r都为1的时候,t
1,1,1
就等于
[0096]
aatp通过位置特异性评分矩阵(pssm)进行aac-pssm特征编码,同时将转移概率矩
阵tpm导入pssm来计算相邻氨基酸残基的关系进行编码,并将aac-pssm生成的20维向量和转移概率组合(tpc)生成的400维向量进行特征组合,合称为aatp。具体的计算公式如下:
[0097][0098][0099]
其中,xj表示由aac-pssm生成的20维特征中的每一维特征,y
i,j
表示由tpc生成的400维特征中的每一维特征。l表示蛋白质序列的长度,p
i,j
表示pssm中第i行,第j列个元素。
[0100]
20种主要的氨基酸可根据疏水性、归一化范德华体积、极性、极化率、二级结构、可溶性、电荷共七种类型的物理化学性质分为三种类别,用class a,class b,class c分别表示。基于理化性质的特征信息可以提升氨基酸序列的表征深度,因此我们将组成、转换和分布(ctd)中的组成(称为ctdc)和转换(称为ctdt)加入cas蛋白的初始特征集(前期称为初始特征集,后期就是12种特征共同构成的特征集)。
[0101]
ctdc根据理化性质按照以下方法进行计算:
[0102][0103]
其中,na分别表示属于某类氨基酸的数量,n是氨基酸序列的长度。
[0104]
ctdt则表示为不同类型残基之间相邻的频率,具体计算为:
[0105][0106]
其中,n
ab
表示是属于classa的残基后紧随属于classb类的残基的出现频率,n
ba
表示属于classb类的残基后紧跟属于classa类的残基出现频率,n表示氨基酸序列的长度。
[0107]
近年来,深度表示学习已被发现是生物序列预测的强大工具。统一表示unirep不同于从前广泛使用的传统氨基酸序列特征表示方法,其使用multiplicative long-/short-term-memory rnns(mlstm),并基于uniref50数据库进行自监督学习,最终生成一个1900维的特征向量,对于一个含有l个氨基酸残基的样本,unirep首先使用one-hot进行编码,并将其嵌入一个l
×
10的矩阵中。随后将矩阵作为mlstm的输入进行unirep特征的计算。
[0108]
本发明包括但不限于上述特征,实际上还可以在此基础上结合其他特征进行识别。
[0109]
s3、通过基于lgbm算法的三步特征空间优化策略对初始特征空间进行特征空间优化;
[0110]
由于基于决策树的分类模型可以根据分裂的总数量和从特征中获得的信息来量化特征的重要性。在三步特征空间优化方法中,首先通过python中lightgbm库中的feature_importances函数计算由s2中十二种特征组成的cas蛋白初始特征集中每一维特
征(不是12种特征,而是12种特征组成的13000维的总特征集中的每一维特征)的特征重要性得分。
[0111]
之后将初始特征集按得分由高到低进行排序并选出初始特征集中得分高于平均得分的特征作为次优特征空间,即:将总特征集的每一维得分相加求平均得分,然后筛选出高于平均得分的特征集作为次优特征空间。随后应用顺序前向搜索方法(顺序前向搜索方法是一个专业术语,就是在特征集中按照特征重要性得分由高到低逐个特征进行累加并计算性能)在次优特征空间中按重要性得分由大到小向特征池中按维添加特征,并将能够获得最大马修斯相关系数(mcc)的特征集作为最优特征空间。
[0112]
s4、引入lgbm算法、rf算法、ert算法、xgboost算法、gbdt算法与步骤s3中得到的最优特征空间进行拟合构建五个基学习模型,并将五个基学习模型输出的置信度得分特征作为第二层模型的输入特征,引入svm算法进行二次建模,构建用于预测cas蛋白的双层预测模型。
[0113]
对于每个基线分类器采用10折交叉验证将cas300随机划分为10个等大小的子集,每次保留一个子集作为验证集,经过十次交叉验证来构建用于训练第二层模型的概率特征。
[0114]
由于产生的概率特征值的分布均在0到1之间,采用了能够更好处理特征值在0到1区间的支持向量机作为第二层分类器。
[0115]
s5、收集cas蛋白的功能域注释文件,利用hmmer算法构建hmm序列数据库。
[0116]
本发明实施例中,共收集了157个crispr-association种子比对。随后使用hmmer软件中的hmmbuild程序从多序列比对结果中构建hmmsprofiles,并通过hmmpress为后续的hmmscan构建了一个profiles数据库。
[0117]
s6、利用s4构建的双层预测模型完成对潜在cas蛋白的初步识别,能够初步筛选出待测蛋白是否为cas蛋白。
[0118]
然后利用hmmer算法对初步识别为cas蛋白的样本在s5中构建的数据库中进行搜索,进行cas蛋白的功能域注释。而hmmer算法能够基于s5构建的数据库对初步筛选出的cas蛋白进行功能域注释,进一步识别cas蛋白的家族。
[0119]
在衡量分类精度时使用准确率(acc)、特异性(sp)、敏感性(sn)、马修斯相关系数(mcc)作为评价指标,具体计算公式如下:
[0120][0121][0122][0123][0124]
其中tp表示预测正确的cas蛋白数量,fp表示预测正确的cas蛋白数量,tn表示预测错误的cas蛋白数量,fn表示预测错误的cas蛋白数量。
[0125]
下面以一组具体实验例对本发明的识别效果作进一步描述。
[0126]
首先,为了评价不同特征的分类性能,在训练集上采用来自基于序列模式、理化性质、进化信息、深度表征学习的十二种特征编码,基于十折交叉验证系统评估了每种特征编码方案在xgboost、gbdt、rf、lgbm、ert分类模型下的性能表现。图2(a)至图2(e)中展示了十二种特征在五种分类模型共六十个模型下的性能表现。从不同特征编码方案的性能评估来看,基于序列模式的asdc编码、基于进化信息的三种编码(aatp、pse-pssm、tri-gram-pssm)以及基于深度表征学习的unirep在不同分类模型下的性能达到了85%以上。以上五种特征编码是识别cas蛋白上性能最优的单一特征,这表明了从多视角进行特征编码的重要性。值得注意的是,基于进化信息的编码在所有分类模型中的准确率均达到了90%以上。具体来说,基于进化信息的三种特征编码(pse-pssm、aatp、tri-gram-pssm)与基于深度表征学习的unirep表现出了相似的性能,且高于基于序列模式(aac、asdc、qso、cksaap、dde、dpc)和基于理化性质的编码(ctdt、ctdc)。这表明虽然基于序列模式和理化性质的编码在识别cas蛋白上也包含着一定的信息并表现出不错的性能,但基于进化信息和深度表征学习编码cas蛋白,能够为cas蛋白分类提供更有效的信息,其相关特征也被认为是建立准确模型的必要条件。
[0127]
为了了解不同特征在最优特征子集中的贡献度,本发明分析了每种特征在最优特征空间的分布和组成。图3(a)和图3(b)的结果中显示,unirep特征占最优特征子集的34.8%,aatp占了39.1%,tri-gram-pssm和pse-pssm分别在最优特征子集中占比17.4%、8.7%。上述四种编码为识别cas蛋白提供了最佳的特征集。此外发现,在最终的特征空间中,基于序列模型信息(qso,asdc,cksaap、aac、dpc、dde)的六种特征和基于理化性质的(ctdt,ctdc)两种特征对cas蛋白的预测没有贡献。从对最佳特征空间组成的分析可以看出,利用包含进化信息的pssm进行特征提取的三种特征全部出现在最终的特征空间。其中,在进化过程中氨基酸残基之间相互突变的平均得分以及应用转移概率矩阵进行pssm转化是成功分类cas蛋白的关键。除此之外,深度表征学习特征对cas蛋白识别的重要性不言而喻。
[0128]
为了评估单一分类模型与集成模型的分类性能,本发明使用最优特征子集将五个基线分类模型与集成模型进行了评估,结果如图4所示,集成模型在acc、sn、sp、mcc上与单一分类模型相比分别超出了1%-3.1%、0.8%-3.3%、0.2%-2.4%、2.6%-6.1%。这也证明了基于stacking策略的集成模型进一步提升了cas蛋白的识别能力。
[0129]
最后,为了评价分类器的泛化能力,本发明使用独立数据集将构建的集成模型与现有方法进行了比较。本发明测试了现有最先进的cas识别方法hmmcas、caspredict。图5详细列出了在独立测试集上的评估结果。发现集成模型在acc、mcc、sn上与caspredict相比分别超出了3.4%、6.5%、6.78%。与hmmcas相比,acc、mcc、sn分别超出了4.24%、10.04%、10.17%。总体而言,stack-cas是基于机器学习的cas蛋白预测模型中最先进的方法,与现有的方法相比能够更准确的发现潜在的cas蛋白。
[0130]
本发明的上述算例仅为详细地说明本发明的计算模型和计算流程,而并非是对本发明的实施方式的限定。对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其它不同形式的变化或变动,这里无法对所有的实施方式予以穷举,凡是属于本发明的技术方案所引伸出的显而易见的变化或变动仍处于本发明的保护范围之列。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1