用于制备脂质体的脂质组合物及其制备方法与流程
1.本发明涉及脂质体技术领域,具体而言,涉及一种用于制备脂质体的脂质组合物及其制备方法。
背景技术:
2.脂质体和脂质纳米粒子的处方中通常含有多个脂质,包括磷脂酰胆碱、胆固醇、磷脂酰乙醇胺以及聚乙二醇化的磷脂酰胆碱或聚乙二醇化磷脂酰甘油。现有的脂质体制备技术是将这些脂质分别称量后,加入到与水混溶的有机溶剂(如无水乙醇)中溶解,而后加入水溶液进行水合,形成脂质体,该方法称为“溶剂注入法”;或者是加入到与水不混溶的有机溶剂(如氯仿或者二氯甲烷)中,溶解为脂质混合物的有机溶液,再通过减压等方法除去有机溶剂后,加入水溶液水合,形成脂质体,该方法称为“薄膜分散法”。
3.采用上述两种方法所制备的脂质体的制剂学性质,包括粒径大小、均匀性、脂质双分子层的结构以及渗透性等,均取决于处方中各个脂质是否可以在水合过程中充分水合并且按照投入比例,在分子水平达到均匀分散。
4.上述两种方法仍存在一些问题。
5.对于溶剂注入法而言:胆固醇是一种晶体,其在有机溶剂如乙醇中的溶解度较低。有机溶剂用量太大时无法形成脂质体,为了保证胆固醇可以在较少的有机溶剂中充分溶解,必须将有机溶剂加热到高于沸点的温度(如当有机有机为乙醇时,需加热至80摄氏度),而后再将胆固醇-有机溶剂的混合体系冷却,加入其他脂质溶解。溶解的温度一般要求在70摄氏度左右。这种方法的缺点在于:需要分步骤、在不同的温度溶解,生产效率低;难以保证各个脂质在分子水平均匀分散,因而各个脂质的浓度和比例与投入值保持一致;高温条件下,磷脂的水解速率大大加快,可能会影响制备得到的脂质体中水解磷脂和游离脂肪酸的含量,并且,这两种磷脂水解的产物会插入到脂质体的脂质膜中,改变膜的排列结构以及渗透性,导致药物泄露。
6.对于薄膜分散法而言:需要使用较大体积的危险有机溶剂,带来环境危害;必须测定脂质体的有机溶剂残留量;与乙醇注入法相比,难以进行工艺放大。
技术实现要素:
7.本发明涉及一种脂质组合物,其为包含符合脂质体制备比例的磷脂和胆固醇的冻干粉,且其中至少部分胆固醇以无定形形式存在。
8.根据本发明的再一方面,还涉及如上所述脂质组合物的制备方法,包括:
9.1)将所述磷脂和所述胆固醇充分溶解在溶剂中;
10.所述溶剂包含药学上可接受的有机溶剂和任选的水,且介电常数为14~19;
11.2)冷冻干燥除去所述溶剂。
12.根据本发明的再一方面,还涉及脂质体的制备方法,包括:
13.将如上所述的脂质组合物分散到水合介质中并任选地装载活性药物;其中所述水
合介质为水或不含有机溶剂的水溶液。
14.本发明所制备得到的脂质组合物(在本发明的其他部分,亦可采用“预制脂质”进行表述)中至少部分胆固醇为无定形,从而极大地提高了脂质组合物在乙醇中的溶解性,采用溶剂注入法时可避免高温溶解,并可获得高浓度的空白脂质体,有利于提高生产效率和批间一致性;同时,该脂质组合物具有极佳的水合能力,因此能有效简化脂质体的制备工艺,缩短制备时间;由于脂质组合物可以充分水合,可保证形成的脂质体具有正确的双分子层结构,从而保证可靠的药物包封率,并提高工艺的稳定性和可靠性。
15.此外,对于含量低的脂质成分,预制脂质能够更好地保持该低含量脂质成分在空白脂质体和载药脂质体中的浓度,进而保证处方中个脂质成分的比例符合预期。由于该预制脂质辅料可以实现脂质成分的充分水合和均匀分散,因此,可以完全不使用有机溶剂,而直接以干粉形式加入到水溶液中,制备为脂质体。对于蛋白、多肽类对有机溶剂类敏感的药物而言,这种预制脂质辅料提供了一种全新的、完全去除有机溶剂的脂质体制备方法,适用于此类药物的装载。这是脂质简单混合物(即将各个脂质简单混合所得)无法实现的。
附图说明
16.为了更清楚地说明本发明具体实施方式或现有技术中的技术方案,下面将对具体实施方式或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图是本发明的一些实施方式,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
17.图1为一个实施例所提供的预制脂质的hplc-cad图谱(a)以及三种脂质的工作曲线(b-d);其中,b为hspc:y=0.6737x+0.7842,r2=0.995;c为胆固醇,y=0.2208x+0.4256,r2=0.998;d为dspe-peg2000:y=0.3364x-0.1718,r2=0.9995;
18.图2为预制脂质s17(表2中编号4)和脂质简单混合物的红外图谱叠加;
19.图3为采用不同溶剂配方制备的两种预制脂质的红外谱图;
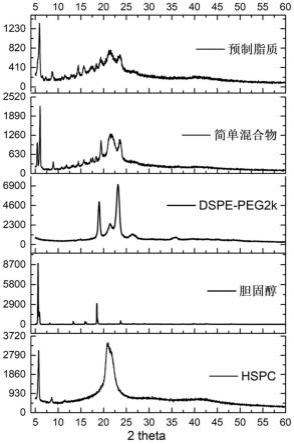
20.图4为单一脂质、简单混合物和预制脂质(s17)的xrd衍射图;
21.图5为脂质简单混合物和预制脂质(s17)的xrd图谱;
22.图6为两种预制脂质的xrd图谱;
23.图7为预制脂质和脂质简单混合物加入硫酸铵溶液中涡旋后的外观;
24.图8为用预制脂质和简单混合物制备的空白脂质体挤出过程中,聚碳酸酯膜表面脂质残留情况;
25.图9为采用预制脂质和脂质简单混合物以直接水合法制备的空白脂质体粒径;
26.图10为预制脂质和简单混合物采用乙醇注入法制备的空白脂质体的粒径;
27.图11为空白脂质体的dsc图;
28.图12为所制备的多柔比星脂质体的冷冻电镜照片;
29.图13为所制备的多柔比星脂质体的dsc图;
30.图14为采用不同脂质原料和制备方法制备的多柔比星脂质体的体外释放曲线;
31.图15为所制备的伊利替康脂质体的冷冻透射电镜照片;
32.图16为采用不同脂质原料制备的伊利替康脂质体的体外释放曲线。
具体实施方式
33.现将详细地提供本发明实施方式的参考,其一个或多个实例描述于下文。提供每一实例作为解释而非限制本发明。实际上,对本领域技术人员而言,显而易见的是,可以对本发明进行多种修改和变化而不背离本发明的范围或精神。例如,作为一个实施方式的部分而说明或描述的特征可以用于另一实施方式中,来产生更进一步的实施方式。
34.除非另有说明,用于披露本发明的所有术语(包括技术和科学术语)的意义与本发明所属领域普通技术人员所通常理解的相同。通过进一步的指导,随后的定义用于更好地理解本发明的教导。本文中在本发明的说明书中所使用的术语只是为了描述具体的实施例的目的,不是旨在于限制本发明。
35.本文所使用的术语“和/或”、“或/和”、“及/或”的选择范围包括两个或两个以上相关所列项目中任一个项目,也包括相关所列项目的任意的和所有的组合,所述任意的和所有的组合包括任意的两个相关所列项目、任意的更多个相关所列项目、或者全部相关所列项目的组合。需要说明的是,当用至少两个选自“和/或”、“或/和”、“及/或”的连词组合连接至少三个项目时,应当理解,在本技术中,该技术方案毫无疑问地包括均用“逻辑与”连接的技术方案,还毫无疑问地包括均用“逻辑或”连接的技术方案。比如,“a及/或b”包括a、b和a+b三种并列方案。又比如,“a,及/或,b,及/或,c,及/或,d”的技术方案,包括a、b、c、d中任一项(也即均用“逻辑或”连接的技术方案),也包括a、b、c、d的任意的和所有的组合,也即包括a、b、c、d中任两项或任三项的组合,还包括a、b、c、d的四项组合(也即均用“逻辑与”连接的技术方案)。
36.本发明中所使用的术语“含有”、“包含”和“包括”是同义词,其是包容性或开放式的,不排除额外的、未被引述的成员、元素或方法步骤。
37.本发明中用端点表示的数值范围包括该范围内所包含的所有数值及分数,以及所引述的端点。
38.本发明中涉及浓度数值,其含义包括在一定范围内的波动。比如,可以在相应的精度范围内波动。比如2%,可以允许
±
0.1%范围内波动。对于数值较大或无需过于精细控制的数值,还允许其含义包括更大波动。比如100mm,可以允许
±
1%、
±
2%、
±
5%等范围内的波动。涉及分子量,允许其含义包括
±
10%的波动。
39.本发明中,涉及“多个”、“多种”等描述,如无特别限定,指在数量上指大于等于2。
40.本发明中,以开放式描述的技术特征中,包括所列举特征组成的封闭式技术方案,也包括包含所列举特征的开放式技术方案。
41.本发明中,“优选”、“更好”、“更佳”、“为宜”仅为描述效果更好的实施方式或实施例,应当理解,并不构成对本发明保护范围的限制。本发明中,“可选地”、“可选的”、“可选”,指可有可无,也即指选自“有”或“无”两种并列方案中的任一种。如果一个技术方案中出现多处“可选”,如无特别说明,且无矛盾之处或相互制约关系,则每项“可选”各自独立。
42.在本发明提及的所有文献都在本技术中引用作为参考,就如同每一篇文献被单独引用作为参考那样。除非和本技术的发明目的和/或技术方案相冲突,否则,本发明涉及的引用文献以全部内容、全部目的被引用。本发明中涉及引用文献时,相关技术特征、术语、名词、短语等在引用文献中的定义也一并被引用。本发明中涉及引用文献时,被引用的相关技术特征的举例、优选方式也可作为参考纳入本技术中,但以能够实施本发明为限。应当理
解,当引用内容与本技术中的描述相冲突时,以本技术为准或者适应性地根据本技术的描述进行修正。
43.本发明第一方面涉及一种脂质组合物,其为包含符合脂质体制备比例的磷脂和胆固醇的冻干粉,且其中至少部分胆固醇以无定形形式存在。
44.在一些实施方式中,所述脂质组合物中的胆固醇中至少50%、60%、70%、80%、90%、95%、96%、97%、98%、99%、99.5%、99.9%的比例以无定形形式存在。
45.根据本发明,“以无定形形式存在”,意味着胆固醇以非晶体存在,基本不发生聚集和重结晶。
46.所述脂质组合物的一个重要特点在于,各脂质成分之间无化学作用,而是通过分子间作用力等物理作用,改善脂质在水和有机溶剂中的分散性和溶解性。
47.该脂质组合物具有极佳的水合能力,因此能有效简化脂质体的制备工艺,缩短制备时间;由于脂质组合物可以充分水合,可保证形成的脂质体具有正确的双分子层结构,从而保证可靠的药物包封率,并提高工艺的稳定性和可靠性。
48.在一些实施方式中,所述胆固醇占所述脂质组合物的38mol%~42mol%,例如39mol%、40mol%、41mol%。
49.磷脂通常为分子内具有由长链烷基构成的疏水基和由磷酸基等构成的亲水基的亲水亲油性物质。作为磷脂,例如可使用磷脂酰胆碱(卵磷脂)、磷脂酰甘油、磷脂酸、磷脂酰乙醇胺、磷脂酰丝氨酸及磷脂酰肌醇这样的甘油脂质、鞘磷脂这样的鞘氨醇磷脂(sphingophospholipid)、心磷脂这样的天然或合成的二磷脂酰系磷脂及它们的衍生物、以及按照常规方法对这些物质进行氢化得到的物质(例如氢化大豆磷脂酰胆碱(hspc))等。磷脂的量在脂质体膜构成成分整体中通常为20mol%以上,优选为40mol%以上,优选为62mol%以下,优选为60mol%以下。
50.在一些实施方式中,所述磷脂包括磷脂酰胆碱。在一些实施方式中,所述磷脂酰胆碱包括二棕榈酰磷脂酰胆碱(dppc),二硬脂酰磷脂酰胆碱(dspc),氢化大豆磷脂(hspc),氢化卵磷脂(hepc)、二肉豆蔻酰磷脂酰胆碱(dmpc)、二月桂酰磷脂酰胆碱(dlpc)、1-硬脂酰-2-棕榈酰磷脂酰胆碱(sppc)、1-豆蔻酰-2-棕榈酰磷脂酰胆碱、1-棕榈酰-2-豆蔻酰磷脂酰胆碱、1-硬脂酰-2-豆蔻酰磷脂酰胆碱、1-棕榈酰-2-豆蔻酰磷脂酰胆碱(pmpc)以及1-棕榈酰-2-硬脂酰磷脂酰胆碱(pspc)中的一种或多种。这些物质中,优选为hspc等进行氢化得到的磷脂、鞘磷脂等。
51.在一些实施方式中,所述磷脂还包括聚乙二醇化磷脂。
52.在一些实施方式中,聚乙二醇化磷脂的含量低于8mol%,例如7mol%、6mol%、5mol%、4mol%、3mol%、2mol%、1mol%、0.5mol%、0.1mol%。
53.在一些实施方式中,所述聚乙二醇化磷脂为聚乙二醇-二硬脂酰磷脂酰乙醇胺(dspe-peg)。
54.dspe-peg可选用例如dspe-peg2000或dspe-peg5000等。
55.作为脂质体膜构成成分,除磷脂、胆固醇等外,还可举出用于脂质体的阴离子性物质和两性离子性物质等。以下进行说明。
56.阴离子性物质可举出二酰基甘油半琥珀酸酯、二酰基甘油半丙二酸酯、二酰基甘油半戊二酸酯、二酰基甘油半己二酸酯、二酰基甘油半环己烷-1,4-二酸、脂肪酸、例如、油
酸、肉豆蔻酸、棕榈酸、硬脂酸、神经酸、山萮酸等,但并不限定于这些。特别优选常温下为固体的饱和脂肪酸,特别优选为棕榈酸、硬脂酸。在本说明书中,常温是指10℃~30℃。上述阴离子性物质相对于脂质体构成成分的整体量的含有比例为0~20mol%,优选为1mol%以上,进一步优选为5mol%以上。
57.两性离子性物质可举出月桂基甜菜碱(月桂基二甲基氨基乙酸甜菜碱)等n-烷基-n,n-二甲基氨基酸甜菜碱;椰油酰胺丙基甜菜碱、月桂酰胺丙基甜菜碱等脂肪酸酰胺烷基-n,n-二甲基氨基酸甜菜碱;椰油酰两性基乙酸钠、月桂酰两性基乙酸钠等咪唑啉型甜菜碱;烷基二甲基牛磺酸等烷基磺基甜菜碱;烷基二甲基氨基乙醇硫酸酯等硫酸型甜菜碱;烷基二甲基氨基乙醇磷酸酯等磷酸型甜菜碱。上述两性离子性物质相对于脂质体构成成分的整体量的含有比例为5~20mol%,优选为1mol%以上,例如5mol%,或例如7mol%。
58.脂质体内还可以含有公知的其它添加剂,例如,作为抗氧化剂,可举出生育酚同系物即维生素e等。另外,修饰脂质体表面的亲水性高分子的脂质衍生物只要不损害脂质体的结构稳定,就没有特别限定,例如可举出聚乙二醇、葡聚糖、普鲁兰、聚蔗糖、聚乙烯醇、合成聚氨基酸、直链淀粉、支链淀粉、甘露聚糖、环糊精、果胶、卡拉胶以及它们的衍生物。其中,优选为聚乙二醇以及聚乙二醇衍生物。亲水性高分子的脂质衍生物的分子量优选为200~5万左右,更优选为1000~1万左右。
59.本发明第二方面涉及如上所述的脂质组合物的制备方法,包括:
60.1)将所述磷脂和所述胆固醇充分溶解在溶剂中;
61.所述溶剂包含药学上可接受的有机溶剂和任选的水,且介电常数为14~19;
62.2)冷冻干燥除去所述溶剂。
63.溶解通常包含两个过程:溶质分子离开其固体聚集体的结构,以及溶质分子的溶剂化。其中,溶剂化过程通常是放热过程(焓值小于零),因此,影响溶质溶解度和溶解快慢的主要因素应为溶质分子获得能量,离开其固体结构的速度。具有规整有序结构的晶体的内能远远高于无定形结构,因此,溶质分子离开晶体结构需要较高的能量,晶体的溶解度小于无定形。在预制脂质的各成分中,胆固醇是一种熔点较高的晶体。因此,我们认为,该预制脂质主要是通过改变了胆固醇(可能还有其他磷脂成分)的物理状态,使其晶体转变为了无定形,从而能够在有机溶剂中达到很高的溶解度,而无需加热到高温,也无需分步溶解。
64.介电常数是一个宏观的参数,它反映出溶剂屏蔽静电作用的能力。介电常数越大,说明这个溶剂越能够有效地屏蔽离子和偶极子两端的吸引和排斥作用,则溶剂化作用越大。发明人意外的发现,在能够充分溶解磷脂和胆固醇的前提下,溶剂的介电常数介于14~19之间(例如14.5、15、15.5、16、16.5、17、17.5、18、18.5)并搭配冷冻干燥,能够使得预制脂质达到上述的无定形状态,并改善其溶解性能。
65.由于冻干工艺制备的固体或者粉末往往具有疏松结构,特别是叔丁醇冰点很低,会在冻干过程中形成大量的针状孔道。这些孔道进一步增加了预制脂质的表面积,因此可以直接加入到水中,快速并且充分地水合。而在有机溶剂中溶解度也大大增加,若叔丁醇含量过低,无法达到保持预制脂质的良好比例。因此,在一些实施方式中,所述溶剂包含60v/v%以上的叔丁醇,例如61v/v%、62v/v%、63v/v%、64v/v%、65v/v%、66v/v%、67v/v%、68v/v%、69v/v%、70v/v%、75v/v%或者更高。
66.在一些实施方式中,所述有机溶剂为包含丙酮、叔丁醇和乙醇的混合体系。
67.在一些实施方式中,所述溶剂包含20v/v%~25v/v%的丙酮,例如21v/v%、22v/v%、23v/v%、24v/v%。
68.在一些实施方式中,所述溶剂包含10v/v%~25v/v%的乙醇,例如15v/v%、20v/v%。
69.在一些实施方式中,所述冷冻干燥的温度为-30℃~-50℃,例如-35℃、-40℃、-45℃。
70.在一些实施方式中,所述冷冻干燥的时间为8h~48h,例如10h、12h、16h、18h、20h、22h、24h、28h、32h、36h、40h、44h。
71.在一些实施方式中,对冷冻干燥后的脂质进行粉碎和/或分装。
72.本发明的第三方面涉及一种脂质体的制备方法,包括:
73.将如上所述的脂质组合物分散到水合介质中并任选地装载活性药物。
74.脂质体可为空白脂质体或载药脂质体。
75.本发明中,水合介质是不包含有机溶剂的水性介质,是指能够分散脂质体膜构成成分的介质,没有特别限定,例如为水,优选为注射用蒸馏水、生理食盐水、葡萄糖水溶液、离子交换水,可以在这些溶液中添加等张剂、缓冲液等,例如,其为蔗糖八硫酸酯三乙胺溶液、硫酸铵溶液、醋酸钙溶液等等。或者,也可以含有作为脂质体内含物质的生理活性物质。
76.在一些实施方式中,所述脂质组合物直接分散到所述水合介质中。
77.由于该预制脂质可以实现脂质成分的充分水合和均匀分散,因此,可以完全不使用有机溶剂,而直接以干粉形式加入到水溶液中,制备为脂质体。
78.在一些实施方式中,先将所述脂质组合物注入到能够与水混溶的有机分散液中溶解,再将所得溶液分散到水合介质中。
79.采用有机溶剂注入法制备脂质体时,无需分步骤溶解,可以将所有脂质成分以预制脂质的形式一次性加入到有机溶剂中,大大简化工艺步骤;并且预制脂质可以保证各脂质成分在有机溶剂中基本完全溶解,并在加入水合介质后,充分水合,从而获得与投入值相近的浓度和比例,从而保证所制备的脂质体的脂质膜具有稳定可控的结构,保证对药物的装载能力,提高工艺的稳定性和可靠性。
[0080]“能够与水混溶的有机分散液”是指所述有机分散液能够微溶、可溶或易溶于水。
[0081]
在一些实施方式中,所述能够与水混溶的有机分散液包括乙醇和/或乙醚,优选乙醇。
[0082]
在一些实施方式中,所述脂质组合物与乙醇的混合体系的温度40℃≤t≤70℃,或40℃≤t≤60℃,或50℃≤t≤60℃,50℃≤t≤70℃,具体的也可以选择如45℃、55℃、65℃。
[0083]
当所制备的脂质体浓度低于50mg/ml时,预制脂质可在低至40摄氏度左右的乙醇中完全溶解。磷脂的水解符合准一级动力学过程。水解速率常数受溶液ph值和温度影响。通常相同ph值时,70℃时磷脂水解速率是40℃时的约10倍。因此,采用有机溶剂注入法制备空白脂质体时,较低的温度有利于保持磷脂的化学稳定性,减少lysopc和脂肪酸的产生,保证脂质体的储存稳定性。当有机溶剂温度在70摄氏度时,该预制脂质的溶解度能够达到约3g/ml以上,完全可以满足工业化大生产中脂质体制备的浓度需要。
[0084]
在一些实施方式中,所述方法还包括用机械方法调控所述脂质体的平均粒径。
[0085]
根据本发明所制备得到的脂质体的粒径可根据本领域技术人员的需要进行调整。
通常采用机械方法调整粒径,在一些实施方式中,所述机械方法包括球磨法、气流粉碎法、高速剪切法、高压均质法、高压挤出法中的一种或多种。粒径的大小(直径)可例如50nm~300nm,例如250nm、200nm、150nm、120nm、110nm、100nm、90nm、80nm、70nm等。
[0086]
所载活性药物包括紫杉烷类、喜树碱类、长春碱类、阿霉素类、环孢菌素类、黄酮类、二氢吡啶类、维a酸类、蒽醌类、挥发油类、鬼白毒素类、嘌呤拮抗剂、嘧啶拮抗剂、叶酸拮抗剂、藤黄酸类、光敏剂类、蛋白类、核酸类中的任一种或多种物质或其衍生物。
[0087]
蛋白类的药物例如抗体或抗体衍生类药物(如adc、抗体核酸偶联药物等)、多肽类激素(如胰岛素、生长激素、促卵泡激素)、细胞因子类药物(如干扰素、粒细胞集落刺激因子、促红细胞生成素、血小板生成素、白细胞介素)、酶(如人尿激酶原、人α葡萄糖苷酶)以及骨形成蛋白2、水蛭素或者其他类型的重组的或天然的蛋白。核酸类药物例如mrna、shrna、mirna、sirna、适配体药物、反义核酸(aso)、激活rna(sarna),或者某些质粒,或携带基因编辑系统(如crispr-cas9系统)的载体。
[0088]
在一些实施方式中,所载活性药物为疫苗活性组分。
[0089]
在一些实施方式中,所在脂质体以组合物的形式被制备得到。因而在制备过程中还任选地包括药物的组装/添加药学上可接受的载体、稀释剂和佐剂等过程。可接受的载体、稀释剂和佐剂对接受者是无毒的并且优选在所使用的剂量和浓度是惰性的,并且包括缓冲剂例如磷酸盐、柠檬酸盐或其他有机酸;抗氧化剂,如抗坏血酸;低分子量多肽;蛋白质,例如血清清蛋白、明胶或免疫球蛋白;亲水性聚合物,如聚乙烯吡咯烷酮;氨基酸,例如甘氨酸、谷氨酰胺、天冬酰胺、精氨酸或赖氨酸;单糖、二糖和其他碳水化合物,包括葡萄糖、甘露糖或糊精;螯合剂如edta;糖醇,如甘露糖醇、木糖醇、赤藓糖醇、麦芽糖醇(maltitol)或山梨糖醇;淀粉,阿拉伯胶(acacia),橡胶,藻酸盐,明胶,磷酸钙,硅酸钙,纤维素,甲基纤维素,微晶纤维素,聚乙烯吡咯烷酮,水,羟基苯甲酸甲酯,羟基苯甲酸丙酯,滑石,硬脂酸镁。
[0090]
下面将结合实施例对本发明的实施方案进行详细描述。应理解,这些实施例仅用于说明本发明而不用于限制本发明的范围。下列实施例中未注明具体条件的实验方法,优先参考本发明中给出的指引,还可以按照本领域的实验手册或常规条件,还可以参考本领域已知的其它实验方法,或者按照制造厂商所建议的条件。
[0091]
下述的具体实施例中,涉及原料组分的量度参数,如无特别说明,可能存在称量精度范围内的细微偏差。涉及温度和时间参数,允许仪器测试精度或操作精度导致的可接受的偏差。
[0092]
实施例1预制脂质的制备
[0093]
1.按照表1的溶剂比例,配制有机溶剂混合物。按照3:1:1的质量比,将氢化豆磷脂(hydrogenated soybean phosphatidylcholine,hspc),胆固醇和聚乙二醇2000-二硬脂酰磷脂酰乙醇胺(peg2000-distearylphosphatidylethanamine,peg2000-dspe)加入到溶剂混合物中,浓度为100mg/ml。
[0094]
2.将各脂质溶液置于40摄氏度水浴中,加热3-5分钟后取出,观察是否澄明。而后,将各脂质溶液置于-40摄氏度冰箱中,冷冻过夜。
[0095]
3.将冷冻后的样品置于冻干机中,冷冻干燥12小时后取出,置于-20度冰箱中密闭保存。
[0096]
4.预制脂质的含量测定:定量称取冻干制备的预制脂质,加入无水乙醇,配制为浓度为1mg/ml的样品。相同方法配制梯度浓度的hspc、胆固醇和peg2000-dspe的乙醇溶液,以及3:1:1质量比的三种脂质的混合溶液,作为对照品。采用高效液相法,用电喷雾检测器(cad)分别测定预制脂质乙醇溶液和对照品溶液中三种脂质的浓度,并计算三者的质量比。结果如表2所示。图1为三种脂质的hplc-cad图谱。
[0097]
表1.用于预制脂质制备的有机溶剂混合物(体积比)
[0098]
溶剂编号丙酮叔丁醇无水乙醇水混合溶剂介电常数脂质溶液加热后是否澄明s15905012.07否s210855012.56否s320755013.54否s4157510013.73否s5207010014.22是s6206020015.58是s72055151021.82否s82050151525.28否s92045152028.74否s102545151525.77否s112550151022.31否s12255515518.85否s133050101022.12否s143045101525.58否s153040102029.04否s16205520519.04否s17206015518.36是s18206515014.9是s19205525016.26是s20255520016.07是s21256015015.39是
[0099]
表2.不同有机溶剂混合物制备的预制脂质中三种脂质的浓度和比例
[0100]
[0101][0102]
按照脂质体配方中三种脂质hspc、胆固醇和dspe-peg2000的质量比要求(分别为58-62%、19-21%和19-21%),表2中编号为5,6,8和9的预制脂质的比例不合格。其余符合预定要求,与对照一致。
[0103]
实施例2预制脂质的物理状态和分子间相互作用表征
[0104]
取表2中9组预制脂质粉末和对照品以及hspc、胆固醇和dspe-peg2000按照质量比3:1:1配制的简单混合物少量,用nicolet is20傅里叶红外(thermo公司,美国)测定其红外光谱。
[0105]
红外光谱结果如图2所示,其中,3400cm-1
为样品中含有的少量水的峰。预制脂质和简单混合物的红外光谱的主要区别在于1700cm-1
~600cm-1
指纹区的差异(分别用浅色和深色圆圈标注)。我们认为,预制脂质中不存在新的化学物质,但是与简单混合物相比,其分子之间存在的作用力不同,因此在乙醇等有机溶剂中易于溶解。
[0106]
图3为采用两种不同的溶剂配方制备的预制脂质的红外谱图。其中,预制脂质s17的三种脂质的浓度和比例符合预期,但预制脂质s12的脂质比例不符合3:1:1的要求,且dspe-peg2000含量偏低。对比s17和s12的红外图谱,二者几乎没有差异,说明s12中脂质的比例不合格,不是由于在制备过程中形成了化学键,或者改变了脂质的分子结构所致。
[0107]
分别取hspc、胆固醇、dspe-peg2000、三者的简单混合物(hspc/胆固醇/dspe-peg2000=3:1:1,w/w),以及表2中编号为9的预制脂质和对照,用d8 advance x射线衍射仪(bruker公司,英国)在5-70度范围内,测定了各样品的衍射行为。
[0108]
如图4所示,胆固醇是一种典型的晶体,具有高强度的晶体衍射峰,dspe-peg2000也具有晶体结构,hspc的晶体结构则较弱。将三者混合并研磨为细粉后,混合物中胆固醇和dspe-peg2000的晶体衍射峰减弱,但仍然存在。对比预制脂质(s17),则发现其中胆固醇和dspe-peg2000的衍射峰几乎消失。图5则更清楚地显示了简单混合物仍存在较为明显的晶体衍射峰(包括胆固醇和dspe-peg2000),而预制脂质(s17)中则非常弱。比较图6中两种预制脂质的xrd图谱,则会发现,s12中仍存在一些胆固醇和dspe-peg2000的晶体衍射峰,而s17中则几乎为无定形。我们认为,这是s12预制脂质中三种脂质的比例不符合预期的主要原因。
[0109]
综上所述,s17所代表的预制脂质的制备方法,主要通过特定介电常数的混合溶剂
改变预制脂质组合物中的分子间作用力,进而改变了脂质(主要为胆固醇)的物理状态,使其晶体转变为了无定形,从而能够在乙醇等有机溶剂中的溶解性质。
[0110]
由于冻干工艺制备的固体或者粉末往往具有疏松结构,特别是叔丁醇冰点很低,会在冻干过程中形成大量的针状孔道。这些孔道进一步增加了预制脂质的表面积,因此可以直接加入到水中,快速并且充分地水合。而在乙醇中溶解度也大大增加,因此,若叔丁醇含量过低,无法达到保持预制脂质的良好比例。
[0111]
实施例3空白脂质体的制备
[0112]
1.采用直接水合法,用预制脂质制备空白脂质体(不含药),并与脂质简单混合物对比
[0113]
称取150mg预制脂质(hspc/胆固醇/dspe-peg2000=3:1:1,w/w);称取90mg hspc,30mg胆固醇和30mgdspe-peg2000(脂质简单混合物);加入3ml硫酸铵溶液(250mm,ph 5.5)中,涡旋振摇1min后观察,可见,预制脂质组气泡较少,而简单混合物组明显有较多气泡(图7)。
[0114]
将两组脂质混悬液置于60摄氏度水浴中,磁力搅拌20min;而后,用挤出器(lipex extruder,evonik)分别用200nm,100nm,80nm和50nm的聚碳酸酯膜逐级过膜挤出。简单混合物组制备的空白脂质体过膜后,200nm挤出膜上明显有白色颗粒状残留,仔细观察有晶体(图8)。由于hspc和dspe-peg2000都是非晶体,所以,挤出膜上的沉淀主要应为胆固醇。由于沉淀堵塞了挤出膜,必须更换才能继续挤出,因此,采用脂质简单混合物制备空白脂质体,在挤出环节需要花费更多时间(表3)。
[0115]
表3.预制脂质和脂质简单混合物直接水合制备空白脂质体挤出环节用时
[0116][0117]
将制备得到的两组空白脂质体,用生理盐水稀释后,采用动态光散射法(zetasizer nanozs90,马尔文公司,英国)测定粒径和粒径分布(图9)。可见,用预制脂质制备的空白脂质体平均粒径更小,并且明显更为均匀。
[0118]
2.采用乙醇注入法,用预制脂质制备空白脂质体(不含药),并与脂质简单混合物对比
[0119]
称取150mg预制脂质(hspc/胆固醇/dspe-peg2000=3:1:1,w/w),全部加入少量无水乙醇中,40℃水浴加热溶解;称取90mghspc,30mg胆固醇和30mgdspe-peg2000(脂质简单混合物);先将胆固醇加入到无水乙醇中,加热溶解,而后冷却,加入其余两种脂质溶解。而后加入3ml硫酸铵溶液(250mm,ph 5.5),于60摄氏度水浴中磁力搅拌20min,水合,制备为脂质体。采用1中相同的方法,过膜挤出;测定挤出后的两组空白脂质体的粒径,结果如图10所示。
[0120]
表4乙醇注入法中用预制脂质和简单混合物制备空白脂质体溶解脂质和挤出步
[0121][0122]
3.用预制脂质制备的空白脂质体的脂质浓度、比例和热力学表征,并与脂质简单混合物制备的空白脂质体进行对比
[0123]
将步骤2和3中用两种方法,以预制脂质和脂质简单混合物为辅料所制备的空白脂质体用10%蔗糖溶液透析过夜,除去外水相中的硫酸铵。用动态光散射法测定了透析后的四组空白脂质体的粒径;用hplc-cad方法,测定了四组空白脂质体中的脂质浓度,并计算了脂质比例。采用差示热扫描方法(microcal capillary dsc,马尔文公司,英国)测定了四组空白脂质体的热力学行为(加热扫描,速度1℃/min;均以10%蔗糖溶液为参比溶液)。
[0124]
透析后的四组空白脂质体的粒径均在100~110nm左右,粒径分布均匀。其中,预制脂质用乙醇注入法制备的脂质体的平均粒径最小(表5)。
[0125]
表5.四组空白脂质体透析后的粒径和pdi(平均值
±
sd,n=3)
[0126][0127]
在脂质浓度和比例方面,脂质简单混合物用直接水合法制备的脂质体中,胆固醇的浓度明显较低,dspe-peg2000的浓度也较低,导致了该脂质体中三种脂质的质量比明显偏离了3:1:1的投入比例(表6)。结合挤出过程中在挤出膜上观察到的固体沉淀,说明用脂质简单混合物为原料,采用直接水合法制备脂质体时,脂质无法充分水合,特别是胆固醇出现了沉淀,因此影响了所制备的脂质体的脂质浓度和比例。而预制脂质采用两种制备方法获得的空白脂质体中三种脂质的浓度和比例均与预制脂质中的比例一致,说明预制脂质保证了各个脂质成分的充分水合和均匀分散。
[0128]
表6.不同原料和制备方法制备的空白脂质体的脂质含量和比例
[0129][0130]
脂质膜的相变行为和热力学参数与脂质浓度和三种脂质的比例密切相关。因此,在定量测定脂质膜中各脂质浓度的基础上,我们还表征了各组空白脂质体的相变行为。如图11所示,四组脂质体的相变温度(tm)均在52度左右,为hspc的相变温度。除了用简单混合物直接水合制备的脂质体外,其余三个空白脂质体样品的dsc曲线完全重合。其中,脂质简单混合物用直接水合法制备的空白脂质体的相变峰明显高窄,而其他三组空白脂质体的相变峰则均扁平。这主要是由于简单混合物用直接水合法制备的脂质体中胆固醇的比例明显较低,因此脂质膜从胶晶相(solid ordered phase)向液晶相(liquid ordered phase)的相变较为显著,呈现出高而窄的相变峰;而其余三组脂质体由于含有较多的胆固醇(摩尔比约40%),处于liquid disordered相,向液晶相的转变较为不显著。
[0131]
将四组空白脂质体的dsc曲线进行定量拟合后所得到的热力学参数值(表7)也显示,简单混合物采用直接水合法制备的脂质体,其脂质膜结构与其余三组不同,具有更高的相变的焓值和一致性(δt
1/2
较小)。而其余三组脂质体的热力学参数与文献报道值相似。说明以预制脂质为原料的制备的脂质体,以及用简单混合物原料,用乙醇注入法制备的脂质体,其脂质膜中三种脂质的含量和比例保持了3:1:1的比例,脂质膜结构相同且与文献报道的空白脂质体一致。
[0132]
表7.各组空白脂质体的热力学参数
[0133][0134]
总结:
[0135]
(1)以预制脂质为原料,可以采用直接水合法和乙醇注入法制备空白脂质体。所得到的空白脂质体的各脂质成分的浓度、比例以及脂质膜的结构均符合预期。
[0136]
(2)与脂质简单混合物相比,直接水合法制备脂质体时,采用预制脂质可以减少搅
拌过程中起泡的产生,保证处方中三种脂质的浓度和比例正确。并完全排除将来大生产挤出过程中脂质(特别是胆固醇)可能存在的沉淀析出,以及带来的溶剂残留的问题。
[0137]
(3)乙醇注入法制备空白脂质体时,预制脂质可以全部加入乙醇中,并在较低温度溶解,比简单混合物易于操作,有利于简化操作流程,并有利于脂质的化学稳定性。与直接水合法相比,可以成为脂溶性药物的选择。
[0138]
实施例4预制脂质用于多柔比星脂质体的制备
[0139]
参照市售多柔比星脂质体(doxil,杨森公司,美国),在实施例3所提供的各空白脂质体中加入多柔比星溶液,于60摄氏度磁力搅拌约20min,进行载药,制备目标药物浓度为2mg/ml,脂质总浓度约16mg/ml的载药脂质体。用动态光散射法测定了载药脂质体的粒径和粒径分布;用树脂吸附法,测定了脂质体的包封率。
[0140]
如表8所示,简单混合物用直接水合法制备的空白脂质体,对多柔比星的包封率非常低(这很可能与水合和挤出过程中胆固醇沉淀析出有关。胆固醇沉淀后,脂质处方改变,渗透性增加,导致载药梯度降低);其余三个空白脂质体对多柔比星的包封率均在99%左右。
[0141]
表8.四组载药脂质体的粒径、粒径分布(平均值
±
sd,n=3)和包封率
[0142][0143]
对于三组包封率在99%的载药脂质体,进一步用冷冻透射电镜观察了脂质体的形貌(图12)。三组脂质体形貌相似,均为咖啡豆形状,内部包含棒状的多柔比星-硫酸铵纳米晶体。
[0144]
采用hplc-cad方法,测定了上述三组包封率在99%的载药脂质体中三种脂质的浓度。三组载药脂质体中各脂质成分的浓度和总脂质浓度均符合预期,并与市售产品doxil一致(表9)。
[0145]
表9.载药脂质体中三种脂质的浓度和脂质总浓度
[0146][0147][0148]
采用dsc表征了这三组包封率高的载药脂质体的热力学行为。测试条件与空白脂质体dsc相同。如图13所示,三组载药脂质体的dsc中,均可观察到两个吸热相变峰:其中,tm在52摄氏度左右的扁平的相变峰为脂质膜的相变;tm在70摄氏度左右的尖锐窄峰则是内水相中多柔比星纳米晶体的熔融峰。这一相变行为也与文献报道的doxil的相变行为一致,说
明采用预制脂质制备的两组空白脂质体装载多柔比星后,所形成的的载药脂质体与doxil具有非常相似的结构(包括脂质膜结构和纳米晶体结构),因此具有相似的热力学参数(表10)。
[0149]
表10.所制备的多柔比星脂质体的热力学参数以及文献报道的doxil和lipodox的热力学参数值(lipodox为印度sun pharma的仿制多柔比星脂质体)
[0150][0151]
三组载药脂质体的体外释放速率研究:
[0152]
以ph 7.4的生理盐水(+-铵根离子)为释放介质,按照1:50体积比,将所制备的三组多柔比星脂质体加入释放介质中,并加入dowex树脂吸附释放出的药物,构造漏槽条件。在不同时间点取样,测定脂质体包裹的药物浓度,计算累积释放百分率,并与doxil的释放行为进行比较。
[0153]
如图14所示,在没有铵根离子的生理盐水中,三组载药脂质体的药物释放速率均十分缓慢,32hr累积释放百分率低于10%,说明三组制剂均保持了稳定的载药性能;在铵根离子的释放促进作用下,三组制剂的释放明显增快,趋势和各时间点的累计释放百分率均很接近,并且非常接近doxil的释放速率。
[0154]
体外释放实验结果表明,所制备的三组多柔比星脂质体释放行为一致,并且与doxil相似。
[0155]
总结:
[0156]
和目前通用的以脂质简单混合物为原料,采用乙醇注入法制备空白脂质体的工艺相比,预制脂质制备的空白脂质体(包括直接水合和乙醇注入法),均可实现稳定、有效的载药,工艺成功率有保证。制备得到的多柔比星脂质体在粒径、形貌、热力学参数以及体外释放行为上与原研可以得到很好的一致。
[0157]
实施例5预制脂质用于伊利替康脂质体的制备
[0158]
1.空白脂质体的制备
[0159]
称取90mg预制脂质(dspc/胆固醇/dspe-peg2000=61.6:38.1:0.3,摩尔比),全部加入少量无水乙醇中,40℃水浴加热溶解;称取67mgdspc,21.8mg胆固醇和1.2mg dspe-peg2000(脂质简单混合物);先将胆固醇加入到无水乙醇中,加热溶解,而后冷却,加入其余两种脂质溶解。而后加入3ml蔗糖八硫酸酯三乙胺溶液(81.25mm,ph 5.5),于60摄氏度水浴中磁力搅拌20min,水合,制备为脂质体;用脂质体挤出器,过膜挤出;测定挤出后的两组空白脂质体的粒径。
[0160]
2.预制脂质制备的空白脂质体的脂质浓度和比例的测定,并与脂质简单混合物制备的空白脂质体进行对比
[0161]
将1中以预制脂质和脂质简单混合物为辅料所制备的空白脂质体用生理盐水透析过夜,除去外水相中的蔗糖八硫酸酯三乙胺。用动态光散射法测定了透析前后的空白脂质体的粒径;用第一组实施例中所描述的脂质的hplc-cad定量,测定了空白脂质体中的脂质浓度,并计算了脂质比例。
[0162]
以预制脂质和脂质简单混合物制备的空白脂质体在透析前后的粒径均在100~110nm左右,粒径分布均匀。(表11)。
[0163]
表11.透析前后空白脂质体的粒径和pdi(平均值
±
sd,n=3)
[0164][0165]
在脂质浓度和比例方面,脂质简单混合物制备的空白脂质体,由于dspe-peg2000的投入量低(制备3ml空白脂质体,仅加入1mg),由于在制备过程中的损失,因此空白脂质体中的dspe-peg2000的测得浓度低于理论值,并由此导致了三种脂质成分比例的改变。而预制脂质所制备的空白脂质体中,三种脂质的浓度和比例则符合预期(表12)。这一结果说明,以预制脂质为辅料,可以避免低含量脂质在制备过程中的损失,从而保证空白脂质体中各脂质成分的比例和浓度。
[0166]
表12.不同原料和制备方法制备的空白脂质体的脂质含量和比例
[0167][0168]
3.预制脂质制备的空白脂质体对伊利替康的装载能力,并与脂质简单混合物制备的空白脂质体进行对比
[0169]
参照市售伊利替康脂质体(onivyde),在各空白脂质体中加入盐酸伊利替康溶液,于55摄氏度磁力搅拌约20min,进行载药,制备目标药物浓度为4.3mg/ml,脂质总浓度约9.2mg/ml的载药脂质体。用动态光散射法测定了载药脂质体的粒径和粒径分布;用电泳法测定了脂质体的zeta电位(zetasizer nanozs90,马尔文公司,英国);用树脂吸附法,测定了脂质体的包封率。两组脂质体的平均粒径和包封率相同(表13)。
[0170]
表13.伊利替康脂质体的粒径、粒径分布,zeta电位(平均值
±
sd,n=3)以及包封率
[0171]
脂质原料粒径(nm)pdizeta电位(mv)包封率(%)简单混合物108.5
±
4.610.053
±
0.014-42.3
±
2.898.5
预制脂质105.4
±
4.020.046
±
0.002-34.3
±
3.299.8
[0172]
两组伊利替康脂质体的形貌基本相同(图15)。内部可见电子密度较高的成分,但是没有明确的精细结构。
[0173]
采用hplc-cad方法,测定了上述两种伊利替康载药脂质体中各脂质成分的浓度。两种脂质体的总脂质浓度均接近市售产品(9.15mg/ml),但是,与空白脂质体中各脂质成分的浓度和比例结果类似,以预制脂质为辅料制备的伊利替康脂质体中三种脂质的浓度和比例符合预期,特别是其中低含量脂质dspe-peg2000的浓度和摩尔比与市售产品一致(分别为0.12mg/ml和0.29%);以脂质简单混合物为辅料制备的脂质体中,dspe-peg2000的浓度和比例均低于市售产品(表14)。
[0174]
表14.载药脂质体中三种脂质的浓度和脂质总浓度
[0175][0176]
表14中,两组伊利替康载药脂质体中dspe-peg2000浓度的差异,也反映在两种脂质体zeta电位的差异上(表13)。简单混合物制备的载药脂质体中,dspe-peg2000浓度低,因此脂质膜中dspc头部磷酸基团的负电荷更易暴露,因此zeta电位的绝对值更高。
[0177]
两组伊利替康载药脂质体的体外释放速率研究:
[0178]
以ph 7.4的生理盐水(+-铵根离子)为释放介质,按照1:50体积比,将所制备的两组伊利替康脂质体加入释放介质中,并加入dowex树脂吸附释放出的药物,构造漏槽条件。在不同时间点取样,测定脂质体包裹的药物浓度,计算累积释放百分率,并与onivyde的释放行为进行比较。
[0179]
如图16所示,在没有铵根离子的生理盐水中,两组载药脂质体的药物释放速率均十分缓慢,24hr累积释放百分率低于10%,说明制剂均保持了稳定的载药性能;在铵根离子的释放促进作用下,两组制剂的释放明显增快,趋势和各时间点的累计释放百分率均很接近,并且非常接近onivyde的释放速率。
[0180]
总结:
[0181]
(1)以dspc/胆固醇/dspe-peg2000=61.6:38.1:0.3(摩尔比)预制脂质为原料,采用乙醇注入法所制备的空白脂质体,与简单混合物所制备的空白脂质体的粒径和分布相近。但是,预制脂质组可以更好的保持各脂质成分,特别是其中低含量脂质成分(dspe-peg2000)的浓度,有利于保持脂质成分的比例,符合投入比例。
[0182]
(2)与目前通用的以脂质简单混合物为原料,采用乙醇注入法制备空白脂质体的工艺相比,用预制脂质制备的空白脂质体,可以稳定、高效地装载伊利替康,所得到的伊利替康脂质体的粒径和形貌与用简单混合物制备的相似。用预制脂质制备的伊利替康脂质体的脂质浓度、比例以及体外释放速率均和市售产品一致。
[0183]
以上所述实施例仅表达了本发明的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对发明专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护
范围。因此,本发明专利的保护范围应以所附权利要求为准,说明书及附图可以用于解释权利要求的内容。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1