基于深度自动编码器的lncRNA-蛋白质相互作用预测方法
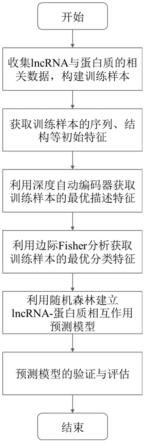
基于深度自动编码器的lncrna-蛋白质相互作用预测方法
技术领域
1.本发明涉及生物信息学领域,特别涉及基于深度自动编码器的lncrna-蛋白质相互作用预测方法。
背景技术:
2.长链非编码rna(long non-coding rna,lncrna)是一类长度大于200个核苷酸、无蛋白质编码能力或编码能力极低的rna转录本,是生物体内重要的分子调控元件。lncrna并不是孤立地存在于生物体内的,它通过传递信号、引导蛋白质定位、集结染色质重构复合物以及诱捕微小rna(mirna)等多种方式发挥其调控功能。由于蛋白质直接参与了生物体内几乎所有的生长发育过程,与蛋白质发生相互作用成为lncrna发挥其调控功能的主要途径。在生物体内,lncrna既可以直接结合特定的蛋白质,也可以招募蛋白质形成rna-蛋白质复合体,与蛋白质之间存在广泛的相互作用。由于蛋白质与lncrna的相互作用在转录后基因调控中起到了重要的作用,如:剪接、信号转导、翻译和复杂疾病的进展,目前国内外研究者对lncrna的关注度持续升温,lncrna-蛋白质相互作用已经成为探索和解析lncrna功能的重要途径。
3.目前预测lncrna-蛋白质相互作用的典型方法有:catrapid、lncpro、rpi-pred和ipminer等。catrapid方法以lncrna与蛋白质的二级结构、氢键作用以及范德华力等理化特性为特征,进而建立基于支持向量机和基于随机森林的预测模型。lncpro方法以lncrna与蛋白质的序列特征之间的关联矩阵为基础,采用线性判别方法进行预测。rpi-pred方法采用序列和三维结构特征,以支持向量机作为分类器预测lncrna-蛋白质相互作用。ipminer方法利用深度学习模型对lncrna和蛋白质的序列组成特征进行多层非线性变换,进而以此为基础建立基于随机森林的预测模型。与其他使用浅层分类器的方法相比,ipminer方法获得了更高的预测准确性。这种性能改进得益于经过多层非线性变换后的特征更具有表征能力,能够准确地描述样本的分布规律。但是,由于lncrna-蛋白质相互作用样本的先验分布(单模或多模)未知,因此在面对对样本分布有严格要求的特征变换方法时ipminer方法的预测准确率并不高。因此目前lncrna-蛋白质相互作用还存在特征表达能力低导致的样本在特征空间的分布区别不大,从而导致lncrna-蛋白质相互作用的预测准确率低的问题。
技术实现要素:
4.本发明目的是为了解决现有lncrna-蛋白质相互作用预测方法还存在特征表达能力低导致的样本在特征空间的分布区别不大,进而导致lncrna-蛋白质相互作用的预测准确率低的问题,而提出了基于深度自动编码器的lncrna-蛋白质相互作用预测方法。
5.基于深度自动编码器的lncrna-蛋白质相互作用预测方法具体过程为:
6.获取待预测的lncrna初始特征和蛋白质初始特征,并将待预测的lncrna初始特征和蛋白质初始特征输入到训练好的lncrna-蛋白质相互作用预测模型中,获得相互作用预测结果;
7.所述训练好的lncrna-蛋白质相互作用预测模型通过以下方式获得:
8.步骤一、获取lncrna-蛋白质相互作用数据文件,并对lncrna-蛋白质相互作用数据文件进行预处理获得原始训练样本集合;
9.步骤二、获取原始训练样本集合中的lncrna的序列及二级结构数据,蛋白质序列、蛋白质骨架结构片段:
10.步骤三、利用原始训练样本集合中的lncrna的序列及二级结构数据,蛋白质序列、蛋白质骨架结构片段分别提取蛋白质特征和lncrna特征,并将获取的蛋白质特征和lncrna特征进行合并获取原始训练样本的初始特征;
11.步骤四、将原始训练样本的初始特征输入到深度自动编码器中进行多层非线性变化获得原始训练样本的初始特征与其重构特征的平均重构误差最小时的编码和解码参数;
12.步骤五、利用边际fisher分析准则和步骤四获得的平均重构误差最小时的最优编码和解码参数对步骤三获得的原始训练样本的初始特征进行处理获得原始训练样本的最优分类特征;
13.所述编码和解码参数为原始训练样本的初始特征与其重构特征的平均重构误差最小时的编码和解码参数;
14.步骤六、利用步骤五获得的原始训练样本的最优分类特征训练lncrna-蛋白质相互作用预测模型获得训练好的lncrna-蛋白质相互作用预测模型。
15.进一步地,所述待预测的lncrna初始特征和蛋白质初始特征通过以下方式获得:
16.s1、获取待预测的lncrna序列和二级结构;
17.所述lncrna的二级结构包括:茎区、发卡、凸起、环、内环;
18.s2、获取待预测蛋白质序列和骨架结构片段;
19.所述骨架结构片段从蛋白质三维结构中抽取;
20.s3、利用待预测的lncrna序列和二级结构提取lncrna初始特征,利用待预测蛋白质序列和骨架结构片段提取蛋白质初始特征:
21.所述利用待预测的lncrna序列和二级结构提取lncrna初始特征,具体为:
22.首先,提取lncrna序列中包含的4-核苷酸聚合体;
23.所述4-核苷酸聚合体共有256种;
24.然后,统计每种lncrna二级结构中包含的每种核苷酸聚合体的数目,对核苷酸聚合体归一化获得lncrna初始特征;
25.所述利用待预测蛋白质序列和骨架结构片段提取蛋白质初始特征,具体为:
26.首先,将每个待预测蛋白质序列中的氨基酸分组,将每组氨基酸用统一字符表示,从而获得多种氨基酸字符串;
27.然后,将蛋白质骨架结构片段分别与每种氨基酸字符串组合,获得复合特征符号;
28.最后,统计每种复合特征符号出现的频率,将复合特征符号进行归一化处理,获得蛋白质初始特征。
29.进一步地,所述将每个待预测蛋白质序列中的氨基酸分组,具体分为如下七组:
30.{a,g,v},{i,l,f,p},{y,m,t,s},{h,n,q,w},{r,k},{d,e}和{c};
31.其中,a是丙氨酸,g是甘氨酸,v是缬氨酸,i是异亮氨酸,l是亮氨酸,f是苯丙氨酸,p是脯氨酸,y是酪氨酸,m是甲硫氨酸,t是苏氨酸,s是丝氨酸,h是组氨酸,n是天冬酰胺,q是
谷氨酰胺,w是色氨酸,r是精氨酸,k是赖氨酸,d是天冬氨酸,e是谷氨酸,c是半胱氨酸。
32.进一步地,所述步骤一中的获取lncrna-蛋白质相互作用数据文件,并对lncrna-蛋白质相互作用数据文件进行预处理获得原始训练样本集合,包括以下步骤:
33.步骤一一、获取lncrna-蛋白质相互作用数据文件;
34.步骤一二、对lncrna-蛋白质相互作用数据文件进行预处理获得原始训练样本集合:
35.首先,对于缺少序列或结构数据的lncrna-蛋白质相互作用对进删除获得删除后的lncrna-蛋白质相互作用对;
36.然后,随机构造与删除后的lncrna-蛋白质相互作用对数目相等的lncrna-蛋白质非作用对;
37.最后,将删除后的lncrna-蛋白质相互作用对、与删除后的lncrna-蛋白质相互作用对数目相等的lncrna-蛋白质非作用对组成原始训练样本集合。
38.进一步地,所述步骤三包括以下步骤:
39.步骤三一、利用原始训练样本集合中蛋白质的序列、蛋白质骨架结构片段提取蛋白质特征v
p
;
40.所述利用蛋白质序列、蛋白质骨架结构片段提取蛋白质特征v
p
的方法与s3中的利用待预测蛋白质序列和骨架结构片段提取蛋白质初始特征相同;
41.步骤三二、利用原始训练样本集中lncrna的序列及二级结构数据提取lncrna特征v
l
;
42.所述利用lncrna的序列及二级结构数据提取lncrna特征v
l
的方法与s3中的利用待预测的lncrna序列和二级结构提取lncrna初始特征的方法相同;
43.步骤三三、将白质特征v
p
与lncrna特征v
l
合并获得原始训练样本的初始特征。
44.进一步地,所述步骤四包括以下步骤:
45.首先,根据每个原始训练样本的初始特征x(i)抽取新特征y(i)和重构特征
46.y(i)=f
θ
(x(i))=s(wx(i)+b)
ꢀꢀ
(1)
[0047][0048]
其中,y(i)是对x(i)进行非线性编码后的特征表示,是对y(i)进行解码后获得的重构特征,θ={w,b}和θ'={w',b'}分别表示编码参数和解码参数,s()是深度自动编码器对特征的作用函数,w是对特征x(i)的作用参数,b是常数项,w、b共同构成了编码参数,w'是对特征y(i)的作用参数,b’是常数项,w
‘
、b’共同构成了编码参数同构成了解码参数;
[0049]
然后,获得x(i)和的平均重构误差最小时的编码和解码参数:
[0050][0051]
其中,是x(i)和之间的重构误差,i∈[1,n]是原始训练样本的标号,n是原始训练样本总数。
[0052]
进一步地,所述步骤五包括以下步骤:
[0053]
步骤五一、构建边际fisher分析准则jmf=sc/s
p
:
[0054]
其中,sc和s
p
分别表示类内紧凑性和类间分离性;
[0055]
步骤五二、利用步骤五一获得的边际fisher分析准则与公式(3)构建提取原始训练样本最优分类特征的目标函数,并对提取原始训练样本最优分类特征的目标函数进行正则化,获得如下目标函数:
[0056][0057]
步骤五三、按照步骤五二获得的目标函数重新训练深度自动编码器,获得最优编码参数θ
*
,从而获得原始训练样本的最优分类特征
[0058]
所述最优编码参数θ
*
利用共轭梯度法获得;
[0059]
所述获得原始训练样本的最优分类特征通过以下方式获得:按θ
*
对原始训练样本的初始特征x(i)进行编码,从而得到原始训练样本的最优分类特征
[0060]
进一步地,
[0061]
其中,sc和s
p
分别表示类内紧凑性和类间分离性,表示与指定的原始训练样本的初始特征属于同类,且是该原始训练样本的初始特征的k
1-近邻样本的集合;表示与指定的原始训练样本的初始特征属于不同类别,但是其k
2-近邻样本的集合,y
(j)
是对x
(j)
进行非线性编码后的特征表示,j是规定范围内任一原始训练样本的特征。
[0062]
进一步地,其中,s
p
是类间分离性,表示与指定的原始训练样本的初始特征属于不同类别,但是其k
2-近邻样本的集合。
[0063]
进一步地,所述lncrna-蛋白质相互作用预测模型的分类器为随机森林算法。
[0064]
本发明的有益效果为:
[0065]
本发明利用深度自编码器学习获得的最优描述特征,准确地刻画了lncrna-蛋白质相互作用样本,提升了特征的表达能力,从而增大样本在特征空间的区别。本发明利用边际fisher分析方法学习lncrna-蛋白质相互作用样本的最优分类特征,提高了lncrna-蛋白质相互作用预测的准确率。本发明的模型泛化性能较好,可应用于识别lncrna-蛋白质之间潜在的相互作用,能够为后续解析lncrna功能以及疾病诊断、药物研发提供很好的先导支持。
附图说明
[0066]
图1为本发明流程图;
[0067]
图2为lncrna-蛋白质相互作用样本的序列和结构特征提取示意图;
[0068]
图3为不同深度自编码器结构对于lncrna-蛋白质相互作用预测性能的影响示意图;
[0069]
图4为不同维度的样本特征对于lncrna-蛋白质相互作用预测性能的影响示意图;
[0070]
图5为初始特征、最优描述特征和最优分类特征对于lncrna-蛋白质相互作用预测性能的影响示意图。
具体实施方式
[0071]
具体实施方式一:本实施方式基于深度自动编码器的lncrna-蛋白质相互作用预测方法具体过程为:获取待预测的lncrna初始特征和蛋白质初始特征,并待预测的lncrna初始特征和蛋白质初始特征输入到lncrna-蛋白质相互作用预测模型中,获得相互作用预测结果,如图1所示;
[0072]
如图2所示,所述待预测的lncrna和蛋白质原始特征通过以下方式获得:
[0073]
s1、获取待预测的lncrna序列和二级结构:
[0074]
从noncode数据库(http://www.noncode.org/index.php)中找出待预测的lncrna并下载这些lncrna对应的核苷酸序列;
[0075]
lncrna的二级结构信息利用rna结构预测工具包viennarna进行抽取;
[0076]
所述lncrna的二级结构信息包括:茎区、发卡、凸起、环、内环;
[0077]
s2、获取待预测蛋白质序列和骨架结构片段:
[0078]
蛋白质的氨基酸序列可以从公开专用数据库uniprot(https://www.uniprot.org/)中下载;蛋白质的骨架结构片段可以从其三维结构数据中抽取,而其三维结构数据可以从公开的蛋白质数据库pdb(ftp://ftp.wwpdb.org/pub/pdb/data/structures/)下载;
[0079]
s3、利用待预测的lncrna序列和二级结构提取lncrna初始特征,利用待预测蛋白质序列和骨架结构片段提取蛋白质初始特征:
[0080]
所述利用待预测的lncrna序列和二级结构提取lncrna初始特征,具体为:
[0081]
首先,lncrna由4种核苷酸组成,提取lncrna序列所包含的4-核苷酸聚合体,获得44=256种核苷酸聚合体特征;
[0082]
然后,统计5种lncrna二级结构中包含的每种核苷酸聚合体的数目,通过对核苷酸聚合体归一化后得到lncrna初始特征;
[0083]
所述利用待预测蛋白质序列和骨架结构片段提取蛋白质初始特征,具体为:
[0084]
首先,按照氨基酸的理化性质,将构成蛋白质序列中的20种氨基酸分为{a,g,v},{i,l,f,p},{y,m,t,s},{h,n,q,w},{r,k},{d,e}和{c}等7组,每组氨基酸用统一的字符表示,则蛋白质的一级序列可以表示为由7种氨基酸字符串。
[0085]
其中,a是丙氨酸,g是甘氨酸,v是缬氨酸,i是异亮氨酸,l是亮氨酸,f是苯丙氨酸,p是脯氨酸,y是酪氨酸,m是甲硫氨酸,t是苏氨酸,s是丝氨酸,h是组氨酸,n是天冬酰胺,q是谷氨酰胺,w是色氨酸,r是精氨酸,k是赖氨酸,d是天冬氨酸,e是谷氨酸,c是半胱氨酸。
[0086]
然后,从蛋白质的三维结构中提取17种蛋白质骨架的结构片段作为蛋白质的结构特征,并将它们分别与7种氨基酸字符串组合,形成119种复合特征符号。最后,统计每种复合特征符号出现的频率,将复合特征符号进行归一化处理后获得蛋白质初始特征。
[0087]
所述训练好的lncrna-蛋白质相互作用预测模型通过以下方式获得:
[0088]
步骤一、获取lncrna-蛋白质相互作用数据文件,并对lncrna-蛋白质相互作用数据文件进行预处理获得原始训练样本集合;
[0089]
步骤一一、获取lncrna-蛋白质相互作用数据文件:
[0090]
从非编码rna相互作用数据库npinter3.0(http://www.bioinfo.org/npinter/)中下载lncrna-蛋白质相互作用数据;
[0091]
步骤一二、对lncrna-蛋白质相互作用数据文件进行预处理获得原始训练样本集合:
[0092]
首先,对于缺少序列或结构数据的lncrna-蛋白质相互作用对进删除;
[0093]
然后,为了使得训练集中的正、负样本数目均衡,随机构造与正样本(lncrna-蛋白质相互作用对)数目相等的lncrna-蛋白质非作用对;
[0094]
最后,将删除后的lncrna-蛋白质相互作用对、与删除后的lncrna-蛋白质相互作用对数目相等的lncrna-蛋白质非作用对组成原始训练样本集合。
[0095]
步骤二、获取原始训练样本集合中的lncrna的序列及二级结构数据,蛋白质序列、蛋白质三维结构及骨架结构片段:
[0096]
从noncode数据库(http://www.noncode.org/index.php)中找出步骤一一中所获的lncrna并下载这些lncrna对应的核苷酸序列;
[0097]
lncrna的二级结构信息利用rna结构预测工具包viennarna进行抽取;
[0098]
所述lncrna的二级结构信息包括:茎区、发卡、凸起、环、内环;
[0099]
蛋白质的氨基酸序列可以从公开专用数据库uniprot(https://www.uniprot.org/)中下载;蛋白质的骨架结构片段可以从其三维结构数据中抽取,而其三维结构数据可以从公开的蛋白质数据库pdb(ftp://ftp.wwpdb.org/pub/pdb/data/structures/)下载。
[0100]
步骤三、利用原始训练样本集合中的lncrna的序列及二级结构数据,蛋白质序列、蛋白质骨架结构片段分别提取蛋白质特征和lncrna特征,并将获取的蛋白质特征和lncrna特征进行合并获取原始训练样本的初始特征,包括以下步骤:
[0101]
步骤三一、利用蛋白质序列、蛋白质骨架结构片段提取蛋白质特征:
[0102]
首先,按照氨基酸的理化性质,将构成蛋白质序列中的20种氨基酸分为{a,g,v},{i,l,f,p},{y,m,t,s},{h,n,q,w},{r,k},{d,e}和{c}等7组,每组氨基酸用统一的字符表示,则蛋白质的一级序列可以表示为由7种字符组成的字符串。
[0103]
然后,从蛋白质的三维结构中提取17种蛋白质骨架的构型片段作为蛋白质的结构特征,并将它们分别与7种氨基酸分组字符相结合,形成119种复合特征符号。最后,统计这些复合特征符号出现的频率,进行归一化处理后作为蛋白质特征v
p
。
[0104]
步骤三二、利用lncrna的序列及二级结构数据提取lncrna特征:
[0105]
首先,lncrna由4种核苷酸组成,提取lncrna序列所包含的4-核苷酸聚合体,获得44=256种核苷酸聚合体特征;
[0106]
然后,统计5种lncrna二级结构中包含的每种核苷酸聚合体的数目,通过对核苷酸聚合体归一化后得到lncrna的特征向量v
l
;
[0107]
步骤三三、将v
p
和v
l
合并形成描述lncrna-蛋白质相互作用原始训练样本的初始特征。
[0108]
步骤四、将原始训练样本的初始特征输入到深度自动编码器中进行多层非线性变化获得原始训练样本的初始特征与其重构特征的平均重构误差最小时的编码和解码参数,
包括以下步骤:
[0109]
步骤四一、将原始训练样本的初始特征输入到深度自动编码器中获得原始训练样本的初始特征与其重构特征的平均重构误差最小时的编码和解码参数:
[0110]
首先,对每一个原始训练样本,都可根据其初始特征x(i)抽取新特征y(i)和重构特征
[0111]
y(i)=f
θ
(x(i))=s(wx(i)+b)
ꢀꢀ
(1)
[0112][0113]
其中,y(i)是利用式(1)对x(i)进行非线性编码后的特征表示,是通过式(2)对y(i)进行解码的结果,θ={w,b}和θ'={w',b'}分别表示编码和解码参数,s()是深度自动编码器对特征的作用函数,w是对特征x(i)的作用参数,b是常数项,w、b共同构成了编码参数,w'是对特征的作用参数,b’是常数项,w
‘
、b’共同构成了编码参数同构成了解码参数。
[0114]
然后,使用描述x(i)和之间的重构误差,当平均重构误差最小时得到最优的编码和解码参数,即
[0115][0116]
其中,是x(i)和之间的重构误差,i∈[1,n]是原始训练样本的标号,n是原始训练样本总数;
[0117]
步骤四二、采用贪心思想逐层训练深度自动编码器,进而得到一个最优的编码参数:
[0118]
在调整深度自动编码器结构时,采用增加隐藏层和缩减特征维度的叠加策略,按隐藏层+1,特征维度-100的步长调整。最终发现包含4个隐藏层,各隐藏层神经元数目为1399-1100-800-500-200-47的深度自动编码器可以获得更好的最佳的预测性能。
[0119]
步骤四三、按照深度自动编码器所获得的最优编码参数对原始训练样本的初始特征进行编码就可以获得原始训练样本的最优描述特征。
[0120]
步骤五、为进一步提升样本特征的区分能力,采用边际fisher分析准则和步骤四获得的原始训练样本的初始特征与其重构特征的平均重构误差最小时的最优编码和解码参数对步骤三获得的原始训练样本的初始特征进行处理获得原始训练样本的最优分类特征,包括以下步骤:
[0121]
步骤五一、构建边际fisher分析准则jmf=sc/s
p
:
[0122][0123][0124]
其中,sc和s
p
分别表示类内紧凑性和类间分离性,表示与指定的原始训练样
本的初始特征属于同类,且是该原始训练样本初始特征的k
1-近邻样本的集合;表示与原始训练样本的初始特征属于不同类别,但是其是该原始训练样本初始特征的k
2-近邻样本的集合,y
(j)
是对x
(j)
进行非线性编码后的特征表示,j是规定范围内任一原始训练样本的特征;
[0125]
k1近邻即给定一个训练数据集,对新的输入实例,在训练数据集中找到与该实例最近邻的k1个实例,这k1个实例的多数属于某个类,就把该输入实例分类到这个类中。
[0126]
k2近邻算法即给定一个训练数据集,对新的输入实例,在训练数据集中找到与该实例最近邻的k1个实例,这k2个实例的多数属于某个类,就把该输入实例分类到这个类中。
[0127]
步骤五二、将步骤五一获得的边际fisher分析准则与式(3)融合起来构建提取原始训练样本最优分类特征的目标函数。为防止过拟合,对该目标函数进行正则化后得到如式(6)所示的新目标函数:
[0128][0129]
步骤五三、按照步骤五二获得的目标函数重新训练深度自动编码器,并求其最优的编码参数,从而得到该原始训练样本的最优分类特征
[0130]
利用共轭梯度法求解最优的编码参数θ
*
,得到最优编码参数θ
*
后,按θ
*
对原始训练样本的初始特征x(i)进行编码,从而得到该原始训练样本的最优分类特征
[0131]
步骤六、利用步骤五获得的最优分类特征训练lncrna-蛋白质相互作用预测模型,并训练好的lncrna-蛋白质相互作用预测模型进行性能评估,包括以下步骤:
[0132]
首先,采用随机森林算法作为分类器,采用前面所获得的原始训练样本的最优分类特征向量训练lncrna-蛋白质相互作用预测模型;
[0133]
然后,利用多种评价指标对训练好的lncrna-蛋白质相互作用预测模型进行性能评估:
[0134]
在性能评估中使用的评价指标包括:精确度(precision)、召回率(recall)、特异性(specificity)、敏感性(sensitivity)、准确率(accuracy)和马修斯相关系数(mcc)。这些指标的具体计算公式如下:
[0135][0136][0137][0138][0139]
[0140][0141]
其中,tp表示预测正确lncrna-蛋白质相互作用的数量,fp表示预测错误的lncrna-蛋白质相互作用数量,tn表示预测正确的非lncrna-蛋白质相互作用数量,fn表示预测错误的非lncrna-蛋白质相互作用数量。
[0142]
实施例:
[0143]
为了评估深度自动编码器结构对预测性能的影响,本实施例建立了层数数目(1,2,3,4,5)不同的深度自动编码器模型,分别计算它们的重建误差。从图3可以明显看出,当层数从2层逐渐增加到5层时,重建误差减小并趋于稳定。当深度自动编码器采用5层结构时,重建误差达到最小,说明5层深度自动编码器结构最为合适。与此同时,本实施例将经由不同网络层学习到的特征对于模型预测性能的影响进行比较,结果如图4所示(图4中每个层节点数的七个的柱形顺序从左到右依次是precision、recall、accuracy、mcc、specificity、sensitivity、auc),经过对比后发现,层节点数为(1100-800-500-200-47)的深度自动编码器能够获得更为准确描述lncrna-蛋白质相互作用的特征子集。
[0144]
为了评估边际fisher分析的有效性,分别利用初始特征、最优描述特征和最优分类特征在测试数据集上预测lncrna-蛋白质相互作用。分析两种预测结果后得到性能如图5所示(图5中每种特征中的七个的柱形顺序从左到右依次是precision、recall、accuracy、mcc、specificity、sensitivity、aue)),经过边际fisher分析后的最优分类特征能够更准确区别lncrna-蛋白质相互作用对与非相互作用对,使预测模型具有更好的性能。
[0145]
为了验证本发明的性能优越性,使用本发明、lncpro、rpi-pred、rpiseq-rf和ipminer等方法在构建的测试数据集上执行lncrna-蛋白质相互作用预测任务,分析它们的预测结果后得到性能如表1所示。从表1可以看出,本发明在所有评测指标上的表现均优于同类方法,具有较好的预测性能。
[0146]
表1与同类方法的性能比较
[0147][0148][0149]
以上提供的本发明的实施例的详细描述并非旨在限制要求保护的本发明的范围,而是仅仅表示本发明的选定实施例。基于本发明的实施例,本领域技术人员在没有做出创造性劳动的前提下所获得的所有其他实施例,都属于本发明保护的范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1