一种基于深度学习的非经典人类白细胞抗原绑定物预测方法
本发明涉及计算生物信息学领域,特别是涉及一种基于深度学习的非经典人类白细胞抗原绑定物预测方法。
背景技术:
1、人类白细胞抗原(hla)是位于第6号染色体的人类组织相容性复合体(mhc)区域的表达产物。人类白细胞抗原密切参与调节人体免疫系统。一般来说,hla基因分为i类、ii类、iii类,而hla i类基因又分成两大类:经典hla(hla-a、hla-b、hla-c)和非经典(hla-e、hla-g、hla-f),目前对于hla基因的研究主要集中在经典hla基因上,近年来,研究表明,非经典hla基因在转录、蛋白质表达和免疫调节中同样起着重要作用。
2、近二十年来,计算方法由于其简单性和有效性而受到更多关注,已经提出了不少于十种用于预测hla绑定物的计算方法,但是大多数计算方法都是基于传统的机器学习(浅层学习)方法,仅限于少量的倾斜样本,模型的泛化能力通常较差,目前只有hlancpred明确用于预测非经典hla i类等位基因的绑定物,hlancpred是一种基于特征工程和传统机器学习的方法,他使用具有不同特征的不同机器学习算法在不同的数据集上构建预测模型,虽然hlancpred获得了相当高的性能,但是hlancpred需要根据不同的数据类型选择不同的机器学习模型,不同的数据类型在同一机器学习模型上的性能好坏不一,这是不方便的。
3、公开号为cn115828152a的专利公开了一种基于图卷积网络的抗癌肽分类方法,包括:s1,获取抗癌肽的训练数据集和测试数据集;s2,将抗癌肽序列构造成以氨基酸节点为顶点的图结构网络并进行编码,获得图结构数据;s3,构建图卷积坍缩池化和残差网络模型,将所述图结构数据输入模型训练;以及,s4,应用训练好的所述模型进行抗癌肽的分类。其中,图卷积坍缩池化和残差网络模型包括堆叠图卷积网络模块、图坍缩池化模块和残差网络模块。上述方案将抗癌肽数据视为一种类似于图结构的数据,利用图卷积神经网络来处理抗癌肽的分类问题,有效区分抗癌肽和非抗癌肽,但是采用单一的卷积神经网络只适用于同种的数据类型,不同的数据类型在同一机器学习模型上的性能好坏不一。
技术实现思路
1、本发明为解决以上背景技术中提到的问题,提供一种基于深度学习的非经典人类白细胞抗原绑定物预测方法,以解决现有技术的问题。
2、本发明采用的技术方案是:
3、一种基于深度学习的非经典人类白细胞抗原绑定物预测方法,包括以下步骤:
4、s1:收集已知的不同等位基因类型的非经典hla肽序列数据集;
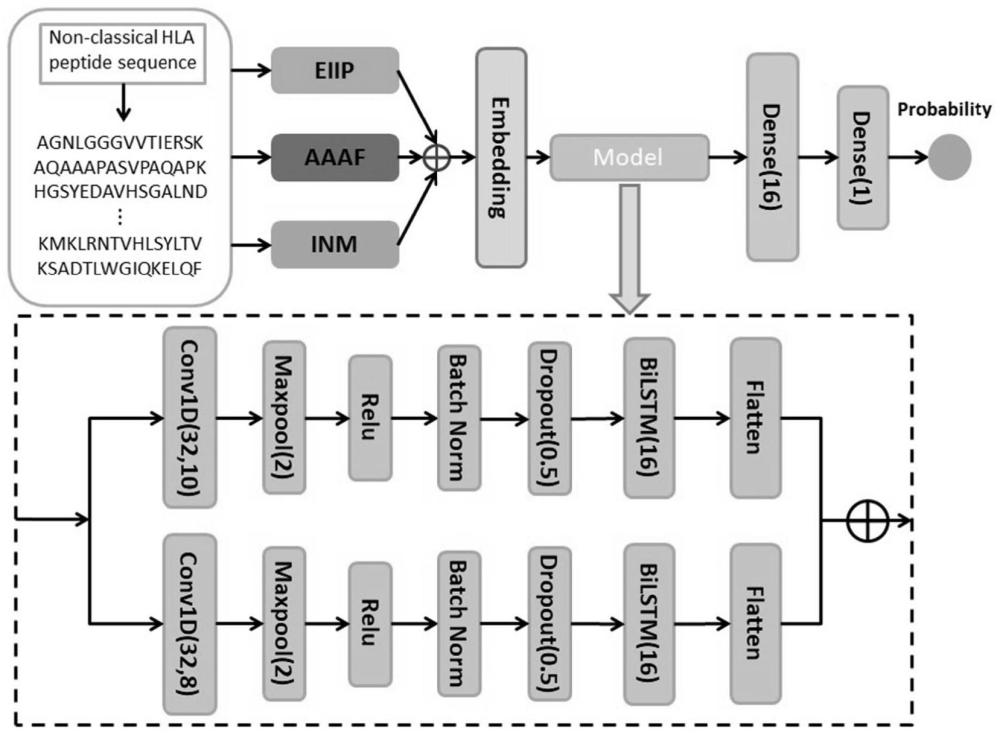
5、s2:分别通过三种特征编码方法电子-离子相互作用势(eiip)、整型数字映射(inm)和累积氨基酸频率(aaaf)提取肽序列的特征表示;
6、s3:将使用三种特征编码方法提取到的特征向量拼接起来,并输送到embedding层中;
7、s4:并行使用不同尺度的卷积神经网络和双向长短时记忆网络构建深度学习模型;
8、s5:对于标注为非经典hla绑定物的肽序列片段视为正样本,将其标签标注为1,否则视为负样本,将其标签标注为0;利用打乱后的数据集对深度学习模型进行有监督训练,通过特征提取方法将打乱后的数据集样本转换为特征向量并输送到embedding层中映射成连续的向量表示,然后输送到搭建好的深度学习模型中进行训练;
9、s6:利用训练好的深度学习模型预测未知的非经典hla绑定物。
10、进一步地,所述步骤s1中将数据集分成两类,分别为平衡数据集和不平衡数据集,在平衡数据集中,每个数据集的正负样本数量相同,而在不平衡数据集中,负样本数量是正样本数量的十倍。
11、进一步地,所述步骤s2中eiip反映了自由电子能的分布,所述其中eiip和inm分别将不同的氨基酸映射成不同的数字向量。
12、进一步地,所述步骤s2中aaaf反映了蛋白质序列中氨基酸的分布密度,aaaf的计算方法包括:假设非经典hla绑定物肽序列s=s1s2…sn,其中n表示肽序列的长度,aaaf的计算公式为其中t(st)的计算公式为
13、其中:j表示肽序列的长度,sj,st表示在某一位置上的氨基酸。
14、进一步地,所述步骤s2中eiip、inm和aaaf特征编码方法将长度为8-15的肽序列编码成15维的向量,对于长度不足15的肽序列,使用0进行填充。
15、进一步地,所述步骤s4卷积神经网络包括卷积和池化操作,其中卷积的计算公式为
16、f(a,b)=∑i∑xc(i,j)i(a+i,b+j),
17、其中:c为卷积核,i表示输入,i和j表示行和列的索引;
18、池化包括最大池化和平均池化,最大池化是返回池化区域的最大值,最大池化计算公式为
19、p(a,b)=maxi,j{f(a+i,b+j)),
20、进一步地,所述步骤s4双向长短时记忆网络包括输入门、遗忘门、输出门以及更新细胞状态,输入门决定哪些信息需要更新,输入门计算公式为
21、it=σ(wi·[ht-1,xt]+bi),
22、其中:xt表示输入,wi表示输入和隐藏状态之间的权重,bi表示偏差,ht-1表示在t-1时刻的隐藏状态;
23、遗忘门决定保留细胞状态的哪些信息,遗忘门计算公式为
24、ft=σ(wf·[ht-1,xt]+bf),
25、其中:wf表示输入和隐藏状态之间的权重,bf表示偏差;
26、输出门决定输出信息,输出门计算公式为
27、ot=σ(wo[ht-1,xt]+bo),
28、其中:wo表示输入和隐藏状态之间的权重,bo表示偏差;
29、候选细胞是一个tanh函数,候选细胞计算公式为
30、
31、其中:wc表示输入和隐藏状态之间的权重,bc表示偏差;
32、输入门和候选细胞组合在一起更新细胞状态,其计算方式为
33、
34、其中:表示逐元素乘法;
35、隐藏状态通过更新细胞状态和输出门更新,其计算公式为
36、ht=ot·tanh(ct);
37、使用双向长短时记忆网络来捕获氨基酸之间的信息,其计算公式为
38、
39、进一步地,所述s4中包含以下内容:两个不同尺度的一维卷积层分别与最大池化层,relu激活层,批标准归一化层,丢弃层,双向长短时记忆网络层和展平层顺序连接,构成了并行网络,将并行网络的两部分输出进行横向连接,输入到两个全连接层,最后一层全连接只使用一个神经元。
40、进一步地,所述s5中具体训练方法为:将随机打乱后的数据集等分成五份,其中四份用于调整模型参数,剩余一份用于预测模型性能,重复五次,确保每一份都用作预测一次。
41、进一步地,在所述s6中包含以下内容:将未知的肽序列通过步骤s2转换为特征向量后,经过步骤s3输送到embedding层,紧接着送入步骤s5训练好的深度学习网络模型中,通过sigmoid函数输出概率值来判断是否为非经典hla绑定物,其中sigmoid函数的计算公式为sigmoid(x)=(1+e-x)-1,其中x表示输入,如果输出概率值大于0.5,则预测肽序列为非经典hla绑定物,否则就不是非经典hla绑定物。
42、与现有技术相比,本发明的有益效果是:
43、本发明利用不同卷积神经网络(convolution neural networks,cnns)和双向长短时记忆网络(bi-directional long short term memory,bi-lstm)组成来搭建神经网络语言模型来提取非经典hla绑定物肽序列和非经典非hla绑定物肽序列的语义特征,进一步增强了氨基酸高水平表达;经过模型训练后,通过sigmoid函数输出概率值来判断是否为非经典hla绑定物,该模型也可以识别未知的序列;在自然语言处理中,句子上下文之间通常包含一定的语义关系,而在生物信息学领域,认为蛋白质序列同样具有该特性,本发明通过实验验证了非经典hla绑定物肽序列中的潜在语义信息,解决了在多类型的数据集中选择指定的模型非常不方便的问题,使用三种不同的特征提取方法,提取了非经典hla肽序列多角度的特征表示,通过embedding层的映射,避免了维度灾难。传统的实验方法费时耗力,通过基于卷积神经网络和双向长短时记忆网络组成的深度学习模型,有效的捕捉了序列之间的语义特征,实现了高精确度的预测性能,是一种高效的预测非经典hla绑定物的深度学习模型方法。
- 还没有人留言评论。精彩留言会获得点赞!