一种氮磷型阻燃剂及其制备方法与流程
本发明涉及阻燃剂技术领域,尤其涉及一种氮磷型阻燃剂及其制备方法。
背景技术:
氮磷系阻燃剂是一种极具发展前途的新型无卤阻燃剂,含有这类阻燃剂的高聚物受热时,表面能够生成一层均匀的碳质泡沫层,起到隔热、隔氧和抑烟的作用,并防止产生熔滴现象,因此,具有良好的阻燃性能。
氮磷系阻燃剂在市场应用中分成小分子量阻燃剂和大分子量阻燃剂两类。小分子量阻燃剂具有生产制造容易、成本低的优点。但其存在分解温度低、热稳定性差等问题,在加工过程中易分解、挥发,不仅使阻燃性能受损,也影响制品的机械性能。大分子量阻燃剂具有性能稳定、阻燃效果好、阻燃剂用量低的优点,但由于分子量大,分子结构紧密使其存在溶解困难、与被阻燃物的相容性差、影响机械强度等缺点。
技术实现要素:
有鉴于此,本发明的目的在于提供一种氮磷型阻燃剂及其制备方法,本发明提供的氮磷型阻燃剂具有优异的溶剂相容性,与合成树脂相容性好,结合力强。
为了实现上述发明目的,本发明提供以下技术方案:
本发明提供了一种具有式i所示结构的氮磷型阻燃剂:
其中,r1为h或-ch3;
r2为
r3为
本发明还提供了上述技术方案所述氮磷型阻燃剂的制备方法,包括以下步骤:
(1)将丙烯酸酯和乙二胺在醇溶剂中发生迈克尔加成反应,得到氨基四元酸酯;
(2)在催化剂的条件下,将所述步骤(1)得到的氨基四元酸酯与多元醇发生酯交换反应,得到氨基四元酸酯多元醇;
(3)将所述步骤(2)得到的氨基四元酸酯多元醇与磷化剂在有机溶剂中发生磷酰化反应,得到氨基四元酸酯多元醇磷酸酯氧磷酰氯;
(4)将所述步骤(3)得到的氨基四元酸酯多元醇磷酸酯氧磷酰氯进行水解反应,得到多元醇磷酸酯酸;
(5)将所述步骤(4)得到的多元醇磷酸酯酸与胺进行成盐反应,得到氮磷型阻燃剂。
优选地,所述步骤(1)中的丙烯酸酯包括丙烯酸甲酯、丙烯酸乙酯、2-甲基丙烯酸甲酯或2-甲基丙烯酸乙酯。
优选地,所述步骤(1)中乙二胺与丙烯酸酯的摩尔比为1:6~10。
优选地,所述步骤(2)中的多元醇包括三羟甲基丙烷、双三羟甲基丙烷、丙三醇、1,3-丙二醇或季戊四醇。
优选地,以乙二胺计,所述步骤(2)中氨基四元酸酯的量以乙二胺计,与多元醇的摩尔比为1:4~8。
优选地,所述步骤(3)中的磷化剂包括三氯氧磷、五氧化二磷或磷酸。
优选地,所述步骤(3)中氨基四元酸酯多元醇的量以乙二胺计,与磷化剂的摩尔比为1:6~9。
优选地,所述步骤(5)中的胺包括三聚氰胺、一乙醇胺、尿素或硫脲。
优选地,所述步骤(5)中多元醇磷酸酯酸的量以乙二胺计,与胺的摩尔比为1:6~9。
本发明提供了一种具有式i所示结构的氮磷型阻燃剂。本发明提供的阻燃剂具有支化结构的多元醇,具备分子间隙大、结构规整。以支化结构的多元醇为中心得到的阻燃剂分子间孔隙多,小分子容易进入;且具有优异的溶剂相容性。另外,本发明提供的氮磷型阻燃剂中存在的氨基,在使其与合成树脂接触时,具有较强的亲和力和结合强度。实施例的数据表明,使用本发明提供的氮磷型阻燃剂对腈纶织物进行整理,在阻燃剂用量200g/l,160℃焙烘150s时,腈纶织物的续燃时间和阴燃时间均为0s,损毁长度3.1cm。
附图说明
图1为实施例1步骤(1)得到的四丙酸酯基乙二胺的红外光谱图;
图2为实施例1步骤(2)得到的四丙酸三羟甲基丙烷酯基乙二胺的红外光谱图;
图3为实施例1步骤(3)得到的四丙酸三羟甲基丙烷酯基乙二胺磷酸酯氧磷酰氯的红外光谱图;
图4为实施例1制备的阻燃剂的红外谱图;
图5为实施例1制备的阻燃剂阻燃腈纶织物燃烧后的外观图片;
图6为实施例2制备的阻燃剂的红外谱图;
图7为实施例2制备的阻燃剂阻燃腈纶织物燃烧后的外观图片。
具体实施方式
本发明提供了具有式i所示结构的氮磷型阻燃剂:
其中,r1为h或-ch3;
r2为
r3为
在本发明的具体实施例中,所述式i所示的结构优选为:
本发明还提供了上述技术方案所述氮磷型阻燃剂的制备的方法,包括以下步骤:
(1)丙烯酸酯和乙二胺在醇溶剂中发生迈克尔加成反应,得到氨基四元酸酯;
(2)在催化剂的条件下,将所述步骤(1)得到的氨基四元酸酯与多元醇发生酯交换反应,得到氨基四元酸酯多元醇;
(3)将所述步骤(2)得到的氨基四元酸酯多元醇与磷化剂在有机溶剂中发生磷酰化反应,得到氨基四元酸酯多元醇磷酸酯氧磷酰氯;
(4)将所述步骤(3)得到的氨基四元酸酯多元醇磷酸酯氧磷酰氯进行水解反应,得到多元醇磷酸酯酸;
(5)将所述步骤(4)得到的多元醇磷酸酯酸与胺进行成盐反应,得到氮磷型阻燃剂。
本发明将丙烯酸酯和乙二胺在醇溶剂中发生迈克尔加成反应,得到氨基四元酸酯。在本发明中,所述丙烯酸酯优选包括丙烯酸甲酯、丙烯酸乙酯、2-甲基丙烯酸甲酯或2-甲基丙烯酸乙酯。在本发明中,所述乙二胺与丙烯酸酯的摩尔比优选为1:6~10,更优选为1:6.5~9,最优选为1:7~8。在本发明中,所述醇溶剂优选包括甲醇。在本发明中,所述迈克尔加成反应的温度优选为10~25℃,更优选为15~23℃,最优选为21~22℃。在本发明中,所述迈克尔加成反应的时间没有特殊的限定,只要迈克尔加成反应液的ph为中性,就可以终止迈克尔加成反应。在本发明中,所述迈克尔加成反应的时间优选为8~50h,更优选为12~48h,最优选为24~36h。
在本发明的实施例中,优选将乙二胺与醇溶剂混合,得到混合液;将丙烯酸酯在1h内滴加到混合液中,进行迈克尔加成反应。本发明对所述乙二胺与醇溶剂的混合方式没有特殊的限定,采用本领域技术人员熟知的混合方式即可,具体的,如搅拌。在本发明中,所述搅拌的转速优选为80~200r/min,更优选为130~150r/min,最优选为150r/min。在本发明中,所述搅拌的时间优选为0.5~2h,更优选为0.5~1h,最优选为0.5h。在本发明中,所述迈克尔加成反应的时间优选以丙烯酸酯滴加完成开始计算。
迈克尔加成反应结束后,本发明优选将迈克尔加成反应产物进行减压蒸馏去除溶剂和多余的原料,得到氨基四元酸酯。在本发明中,所述减压蒸馏的压力优选为3.5~12kpa,更优选为5.5~9.5kpa,最优选为8.5~9.5kpa。在本发明中,所述减压蒸馏的温度优选为20~50℃,更优选为25~45℃。在本发明中,所述减压蒸馏的时间优选为1~3h,更优选为1~2h,最优选为1.2~1.5h。
在本发明中,所述迈克尔加成反应优选在搅拌的条件下进行。在本发明中,所述搅拌的转速优选为80~200r/min,更优选为130~150r/min,最优选为130~140r/min。
在本发明中,迈克尔加成反应能够形成以乙二胺为核心的初步支化结构,为最终氮磷型阻燃剂的大空间结构提供基础。
得到氨基四元酸酯后,本发明在催化剂的条件下,将氨基四元酸酯与多元醇发生酯交换反应,得到氨基四元酸酯多元醇。在本发明中,所述多元醇优选包括三羟甲基丙烷、双三羟甲基丙烷、丙三醇、1,3-丙二醇或季戊四醇。在本发明中,所述氨基四元酸酯的量以乙二胺计,与多元醇的摩尔比优选为1:4~8,更优选为1:5~7,最优选为1:5.5~6。在本发明中,所述催化剂优选包括甲醇钠、碳酸钾、阳离子交换树脂或硫酸氢钠。在本发明中,所述催化剂的用量优选为氨基四元酸酯与多元醇总质量的0.5~3%,更优选为1~2.5%,最优选为1.5~2%。
在本发明中,所述酯交换反应的温度优选为80~110℃,更优选为85~100℃,最优选为90~95℃。在本发明中,所述酯交换反应的时间优选为8~12h,更优选为9~11h,最优选为9.5~10h。
在本发明中,所述酯交换反应优选在搅拌的条件下进行。在本发明中,所述搅拌的转速优选为80~200r/min,更优选为100~180r/min,最优选为120~130r/min。
酯交换反应结束后,本发明优选将酯交换反应产物进行减压蒸馏,去除酯化反应产物中的醇类,得到氨基四元酸酯多元醇。在本发明中,所述蒸馏的温度优选为70~120℃,更优选为80~100℃,最优选为90℃。在本发明中,所述减压蒸馏的压力优选为3.5~12kpa,更优选为5.5~10kpa,最优选为8.5~9.5kpa。在本发明中,所述蒸馏的时间优选为1~3h,更优选为1.2~2h,最优优选为1.5h。
在本发明中,以多元醇为原料,发生酯交换反应后,在氨基四元酸酯中的以乙二胺为核心的支化分子外周形成多个羟基取代基,为后续磷酸酯化反应创造条件;同时在阻燃剂中引入羟基基团,保证了阻燃剂的溶剂相容性和与合成树脂的相容性。
得到氨基四元酸酯多元醇后,本发明将氨基四元酸酯多元醇与磷化剂在有机溶剂中发生磷酰化反应,得到氨基四元酸酯多元醇磷酸酯氧磷酰氯。在本发明中,所述磷化剂优选包括三氯氧磷、五氧化二磷或磷酸。在本发明中,所述氨基四元酸酯多元醇的量以乙二胺计,与磷化剂的摩尔比优选为1:6~9,更优选为1:6.5~8,最优选为1:7~7.5。在本发明中,所述有机溶剂优选包括1,4-二氧六环。在本发明中,所述有机溶剂的用量(按质量比反应物:溶剂)优选为6~1:1,更优选为5~2:1,最优选为4~3:1。
在本发明中,所述磷酰化反应的温度优选≤90℃,更优选为50~70℃。在本发明中,所述磷酰化反应的时间优选为8~20h,更优选为10~18h,最优选为12~15h。
在本发明中,所述磷酰化反应优选在搅拌的条件下进行。在本发明中,所述搅拌的转速优选为60~200r/min,更优选为80~140r/min,最优选为90r/min。
在本发明中,优选对磷酰化反应产生的氯化氢进行尾气吸收。
磷酰化反应结束后,本发明优选对磷酰化反应产物进行减压蒸馏除去有机溶剂和未反应的磷化剂以及生成的氯化氢,得到氨基四元酸酯多元醇磷酸酯氧磷酰氯。在本发明中,所述减压蒸馏的温度优选为70℃;所述减压蒸馏的压力优选为12kpa;所述减压蒸馏的时间优选为2h。
得到氨基四元酸酯多元醇磷酸酯氧磷酰氯后,本发明将氨基四元酸酯多元醇磷酸酯氧磷酰氯进行水解反应,得到多元醇磷酸酯酸。在本发明中,所述氨基四元酸酯多元醇磷酸酯氧磷酰氯的量以乙二胺计,与水的摩尔比优选为1:4~6,更优选为1:4.5~5.5,最优选为1:5。在本发明中,所述水解反应的温度优选为30~60℃,更优选为35~50℃,最优选为40~45℃。在本发明中,所述水解反应的时间优选为4~8h,更优选为4.5~7h,最优选为5~6h。
得到多元醇磷酸酯酸后,本发明将多元醇磷酸酯酸与胺进行成盐反应,得到氮磷型阻燃剂。在本发明中,所述胺优选包括三聚氰胺、一乙醇胺、尿素或硫脲。在本发明中,所述多元醇磷酸酯酸的量以乙二胺计,与胺的摩尔比优选为1:6~9,更优选为1:6.5~8,最优选为1:7~7.5。在本发明中,所述成盐反应的温度优选≤90℃,更优选为60℃。在本发明中,所述成盐反应的时间优选为8~20h,更优选为12~18h,最优选为14~16h。
在本发明中,所述成盐反应优选在搅拌条件下进行,所述搅拌的转速优选为60~120r/min,更优选为70~100r/min,最优选为80r/min。
在本发明中,所述成盐反应结束后,本发明优选将成盐反应产物进行固液分离,对得到的滤渣进行洗涤和干燥,得到所述氮磷型阻燃剂。在本发明中,所述固液分离优选为过滤。在本发明中,所述洗涤的药剂优选为去离子水。在本发明中,所述干燥的温度优选为80℃,所述干燥的时间优选为10h。
下面结合实施例对本发明提供的氮磷型阻燃剂及其制备方法进行详细的说明,但是不能把它们理解为对本发明保护范围的限定。
实施例1
结构式如下式所示的氮磷型阻燃剂的制备方法,包括以下步骤:
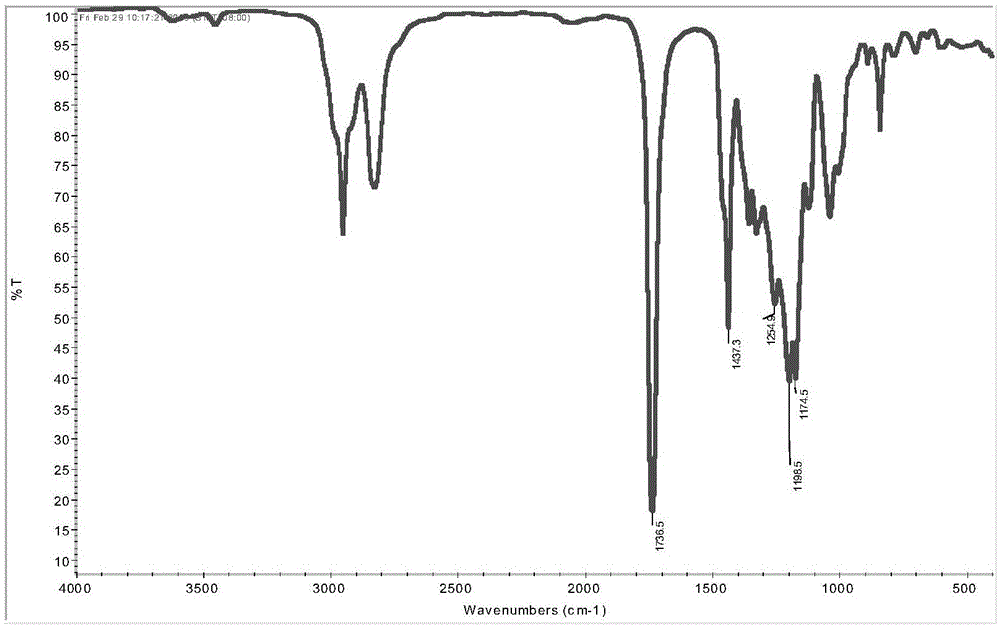
(1)将乙二胺和甲醇于带有磁力搅拌子、回流冷凝管和温度计的四口烧瓶中搅拌混合10min,得到混合液,将丙烯酸甲酯于1h内滴加到混合液中;于22℃进行迈克尔加成反应,当迈克尔加成反应液的ph值接近7.0时,停止反应;迈克尔加成反应结束后,于40℃,8.5kpa下减压蒸馏,除去甲醇和过量的丙烯酸甲酯,得四丙酸酯基乙二胺;其中,乙二胺和丙烯酸甲酯的摩尔比1:8;采用红外光谱法研究步骤(1)所得四丙酸酯基乙二胺的结构,结果如图1所示。从图1可以看出:在2800~3100cm-1处有甲基和亚甲基的吸收峰,1437cm-1为甲基弯曲振动吸收峰,在1736cm-1为酯键的特征吸收峰,842cm-1为=ch2的特征峰;与期望目的产物结构基团相符。
(2)以乙二胺计,将四丙酸酯基乙二胺与三羟甲基丙烷按摩尔比1:6混合,加入四丙酸酯基乙二胺与三羟甲基丙烷总质量2%的甲醇钠,于90℃下进行酯交换反应10h,酯交换反应结束后,于90℃,9.5kpa下减压蒸馏,除去酯交换反应生成的甲醇,得到四丙酸三羟甲基丙烷酯基乙二胺;采用红外光谱法研究步骤(2)所得四丙酸三羟甲基丙烷酯基乙二胺的结构,结果如图2所示。从图2可以看出:在3312cm-1处为-oh的伸缩振动吸收峰,1023cm-1处为-oh的弯曲振动吸收峰,2800~3000cm-1的是甲基和亚甲基的伸缩振动吸收峰,1462cm-1为甲基和亚甲基的弯曲振动吸收峰,在1723cm-1为酯键的特征吸收峰,与期望目的产物结构基团相符。
(3)以乙二胺计,将四丙酸三羟甲基丙烷酯基乙二胺与三氯氧磷按摩尔比1:6与有机溶剂1,4-二氧六环(反应物:溶剂按质量比2:1)混合,于50℃下进行磷酰化反应10h;磷酰化产生的氯化氢直接进行尾气吸收;磷酰化反应结束后,于70℃、9.5kpa压力下减压蒸馏除去液体中未反应的三氯氧磷和1,4-二氧六环,得到四丙酸三羟甲基丙烷酯基乙二胺磷酸酯氧磷酰氯;采用红外光谱法研究所述步骤(3)得到的四丙酸三羟甲基丙烷酯基乙二胺磷酸酯氧磷酰氯的结构,结果如图3所示。从图3可以看出:在2975cm-1处为甲基的伸缩振动吸收峰,1465.2cm-1为亚甲基的弯曲振动吸收峰,在1739.9cm-1为酯键的伸缩振动峰,在1044cm-1为叔胺的伸缩振动峰,在1294cm-1为o=p的特征吸收峰,在462cm-1为p-cl的特征吸收峰,与期望目的产物结构基团相符。
(4)以乙二胺计,将四丙酸三羟甲基丙烷酯基乙二胺磷酸酯氧磷酰氯与去离子水按摩尔比1:6混合,于75℃进行水解反应4h,得到四丙酸三羟甲基丙烷酯基乙二胺磷酸酯磷酸;
(5)以乙二胺计,将四丙酸三羟甲基丙烷酯基乙二胺磷酸酯磷酸与三聚氰胺按摩尔比1:8混合,于60℃进行成盐反应时间7h;成盐反应结束后,将成盐反应液过滤、清水洗涤和干燥,得到磷酸酯盐阻燃剂;采用红外光谱法研究所述步骤(5)得到的磷酸酯盐阻燃剂的结构,结果如图4所示。从图4可以看出:在3400~3100cm-1有伯胺盐和-nh2的强吸收峰,在1552cm-1和1327cm-1处有nh3+的反对称和对称弯曲振动吸收峰,在1677cm-1处有c=n伸缩振动强吸收,由此可证明三聚氰胺盐结构的存在。在1096cm-1、1009cm-1是p-o-c振动吸收峰;与期望目的产物结构基团相符。
实施例1中(1)~(5)步骤的反应原理如下所示:
步骤(1):michael加成反应
步骤(2):酯交换反应
步骤(3):酯交换产物磷酰化反应
步骤(4):水解反应
步骤(5):水解产物成盐反应
将实施例1制备的阻燃剂用于整理腈纶织物,在阻燃剂用量分别为150g/l、200g/l时,浴比为1:10,经两浸两轧,轧液率在90~100%之间,然后在70℃预烘180s,在160℃焙烘150s。参照gb/t5455-1997《织物燃烧性能试验垂直法》标准,用yg2815a型织物阻燃性能测试仪平行测试3次,取平均值。织物阻燃性的结果如表1所示;织物燃烧后状态如图5所示。从表1可以看出:实施例制备的阻燃剂对腈纶织物具有优异的阻燃性。
表1阻燃剂整理腈纶数据表
实施例2
结构式如下式所示的氮磷型阻燃剂的制备方法,包括以下步骤:
(1)将乙二胺和甲醇于带有磁力搅拌子、回流冷凝管和温度计的四口烧瓶中搅拌混合10min,得到混合液,将甲基丙烯酸甲酯于1h内滴加到混合液中;于22℃进行迈克尔加成反应,当迈克尔加成反应液的ph值接近7.0时,停止反应;迈克尔加成反应结束后,于50℃,8.5kpa下减压蒸馏,除去甲醇和过量的甲基丙烯酸甲酯,得四异丁酸甲酯基乙二胺;其中,乙二胺和丙烯酸甲酯的摩尔比1:8;
(2)以乙二胺计,将四异丁酸甲酯基乙二胺与三羟甲基丙烷按摩尔比1:8混合,加入四异丁酸甲酯基乙二胺与三羟甲基丙烷总质量2%的甲醇钠,于90℃下进行酯交换反应10h,酯交换反应结束后,于90℃,8.5kpa下减压蒸馏,除去酯交换反应生成的甲醇,得到四异丁酸三羟甲基丙烷酯基乙二胺;
(3)以乙二胺计,将四异丁酸三羟甲基丙烷酯基乙二胺与三氯氧磷按摩尔比1:3与有机溶剂1,4-二氧六环混合,于70℃下进行磷酰化反应8h;磷酰化反应结束后,于70℃、12kpa压力下减压蒸馏除去液体中的1,4-二氧六环,得到四异丁酸三羟甲基丙烷酯基乙二胺磷酸酯氧磷酰氯;
(4)以乙二胺计,将四异丁酸三羟甲基丙烷酯基乙二胺磷酸酯氧磷酰氯与去离子水按摩尔比1:6混合,于75℃进行水解反应4h,得到四异丁酸三羟甲基丙烷酯基乙二胺磷酸酯磷酸;
(5)以乙二胺计,将四异丁酸三羟甲基丙烷酯基乙二胺磷酸酯磷酸与三聚氰胺按摩尔比1:6混合,于60℃进行成盐反应时间7h;成盐反应结束后,将成盐反应液过滤、清水洗涤和干燥,得到磷酸酯盐阻燃剂。
采用红外光谱研究制备得到的磷酸酯盐阻燃剂的结构,结果如图6所示。从图6可以看出:在3400~3100cm-1有伯胺盐和-nh2的强吸收峰,在1506.8cm-1和1338.1cm-1处有nh3+的反对称和对称弯曲振动吸收峰,在1684.6cm-1处有c=n伸缩振动强吸收,由此可证明三聚氰胺盐结构的存在。在1077cm-1、981.7cm-1是p-o-c振动吸收峰,在1034cm-1为叔胺的伸缩振动峰,在1211.8cm-1为酯基的伸缩振动峰;与期望目的产物结构基团相符。
将实施例2制备的阻燃剂整理腈纶织物,阻燃剂的用量分别为150g/l和200g/l时,在浴比为1:10,经两浸两轧,轧液率在90~100%之间,然后在70℃预烘180s,在160℃焙烘150s。参照gb/t5455-1997《织物燃烧性能试验垂直法》标准,用yg2815a型织物阻燃性能测试仪平行测试3次,取平均值。织物阻燃性的结果如表2所示;织物燃烧后状态如图7所示。
表2阻燃剂整理腈纶数据表
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!