用于氙回收的低温吸附方法与流程
本发明整体涉及一种用于从低温液体或气流中回收氙的吸附方法,其中吸附床与含氙的液体或气流接触并从气流中选择性地吸附氙。将吸附床保持在进料上,直到流出物氙浓度等于或大于入口氙浓度的70%,这使得在使用温度摆动方法进行再生之前能够深度排斥其它流组分。在再生之前以这种方式操作吸附床使得能够从吸附床产生高纯度产物,并且进一步使得氧气能够安全地用作吹扫气体,即使在烃共同存在于进料流中的情况下也是如此。
背景技术:
将吸附床保持在进料上,直到床的出口处的xe浓度大于或等于入口浓度的90%,这使得能够实现在氧气可安全地用作温度摆动吸附方法的吹扫流体的条件下操作的方法。此外,通过这种方式,可在其它气体可共吸附的压力和温度条件下操作床。与现有技术相比,这有助于扩展本发明方法可用的方法条件范围。
例如,在shino等人的美国专利5,039,500中,公开了一种从空气分离单元的主冷凝器中用液态氧产生高纯度氙的方法。在shino等人的方法中,含氙、氪和烃的液态氧流首先被气化,然后在预选的温度和压力下与吸附剂接触,以便在吸附剂上吸附氙,但不吸附包含在氧流中的氧、氪或烃。使用吹扫气体并通过加热使吸附剂再生。通过其它单元操作可增强基本吸附方法,以提高氙的纯度,这些单元包括固气分离塔、催化剂塔、水分和co2去除塔等。该方法的缺点是,来自低温设备的液体进料流在与吸附床接触之前必须转化为气流。此外,根据权利要求1,吸附方法必须在预选的温度和压力条件下操作,使得氙被吸附而氪、烃和氧不被吸附。这限制了该方法的操作条件。然而,根据实施方案1(第3栏,第20-48行)可以看出,这种识别压力和温度条件的方法(其中氙被吸附而氧、氪和烃不被吸附)只是部分成功。在该实施方案中,使硅胶吸附剂接触,直到在-170℃下用在氧基质中含31ppm氙、70ppm氪和38ppm甲烷以及低浓度的其它烃的气化流实现xe穿透。在将气体加热至120℃以使吸附剂再生后,流出物浓度变为1.4%的氙、0.14%的氪、0.066%的烃和余量的氧。事实是氪和烃以与氙相同的方式显著富集超过其进料浓度,表明在用于吸附进料步骤的条件下这些组分也被吸附,这与所要求保护的方法明显矛盾。因此,由于其它组分、特别是烃组分发生共吸附并且它们通过吸附发生富集,后来的实施方案可能使用催化剂塔来去除这些烃,然后使用co2和水分去除塔来去除催化剂上烃燃烧的产物。相比之下,在本发明的吸附方法中,进料流可以是液相或气相,并且在吹扫和加热步骤中,使用该进料流来回收氙产物,氪和烃的浓度远远小于它们在进料中的浓度。在shino等人的技术中,如实施方案1中所阐明的,在用于使吸附床再生的加热步骤中,进料中的38ppm甲烷和低浓度的其它烃变为0.066%或660ppm。这是进料中烃浓度的大约17倍的富集。
同样是shino等人的美国专利4,874,592公开了一种吸附-解吸方法,其中通过连续的吸附和解吸阶段从排出的液态氧气流中浓缩氙,并且其中从第一吸附阶段后回收的氙气流中催化去除烃。根据实施例1和实施例2并且如美国专利4,874,592的图1和图2所示,将来自精馏塔的含排出的稀有气体的流引入第一吸附塔,其中允许能够选择性地吸附氙的硅胶吸附剂饱和。通过降低压力并且通过加热塔来收集来自该第一吸附塔的产物流。该产物流含有富集浓度高于进料组成的氙、氪和烃的混合物。使用催化剂单元操作以及随后的二氧化碳和水去除塔来去除烃,然后使用第二吸附塔来进一步提高稀有气体产物的纯度。如该专利的实施例1中所阐明的,排出液态氧流会产生到吸附系统的气态氧进料流。如上所述,本发明的吸附方法与液体或气体进料相容,因此不需要排出液态氧的步骤。此外,操作本发明的方法使得可避免烃的富集超过它们在进料流中的浓度,因此不需要如现有技术中所述的催化氧化的步骤。
golden等人的美国专利6,658,894公开了一种通过使用与li和ag交换的x型沸石选择性地吸附氙和/或氪来从含氧的气流中回收氙或氪中的至少一种的方法。根据实施例7,其示出了golden等人的方法中的关键步骤,即使含17ppm氙、95ppm甲烷和10ppm一氧化二氮的液态氧流通过硅胶床,一氧化二氮在该硅胶床中被去除。将不含一氧化二氮的流出物蒸发至113k,并且将一部分该气流送至含与锂和银交换的x型沸石的床中。在运行190分钟后检测到甲烷的穿透,而在运行1400分钟后,没有发生氙的穿透。此时,停止进料步骤并在113k下使用氮吹扫气体开始再生。根据golden等人专利的图4中的数据,解吸过程中的甲烷浓度增加至最大值8000ppm-9000ppm。通过进一步加热吸附床来收集氙产物。golden等人的技术的关键特点是:
·使用与li和ag交换的x型沸石。
·将吸附方法操作至未观察到氙的穿透的点。
·在氮气下解吸,其中出口处的甲烷浓度显著超过进料流中的甲烷浓度(95ppm与8000ppm-9000ppm)。
在本发明的方法中,解吸过程中的甲烷水平未表现出在golden等人呈现的数据中观察到的这种富集行为。此外,本发明的方法可在液相中操作,并且不需要li和agx型沸石。
golovko的美国专利3,971,640公开了一种用于从空气流中回收氪-氙浓缩物的吸附方法。在golovko的方法中,使含氪、氙和烃的混合物的气态气流在90k-110k下通过孔开口为
技术实现要素:
描述了一种用于从低温液体或气流中回收氙的吸附方法,其中吸附床与上述含氙的液体或气流接触并从该气流中选择性地吸附氙。操作吸附床以至少接近氙的完全穿透,以使得在使用温度摆动方法进行再生之前能够深度排斥其它流组分。在再生之前操作吸附床以接近氙的完全穿透使得能够从吸附床产生高纯度产物,并且进一步使得氧气能够安全地用作吹扫气体,即使在烃共同存在于进料流中的情况下也是如此。
附图说明
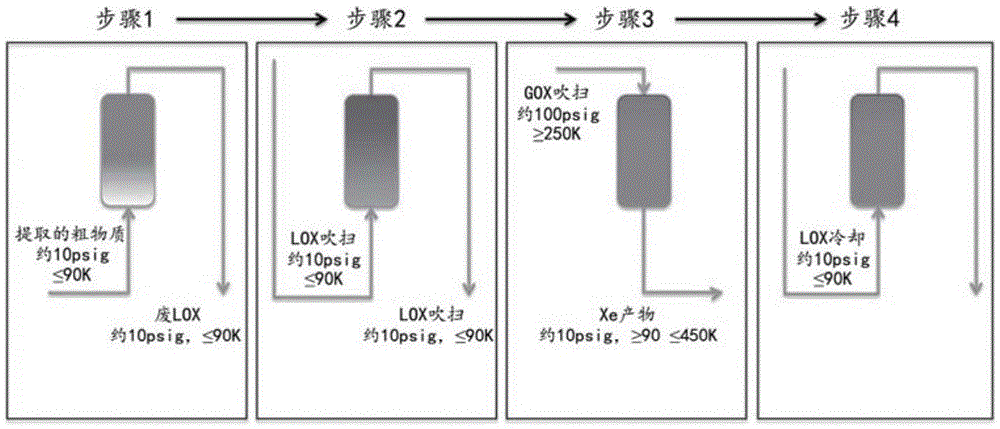
图1是示出分别在步骤1和步骤2中用液体进料和吹扫进行1-床吸附方法的方法步骤的示意图。
图2示出了在进料(顶部)和解吸(底部)期间吸附床的出口处的ch4和xe的浓度曲线。
图3示出了在提取步骤中吸附床的出口处的ch4、xe、kr和n2o的浓度对时间的曲线。
图4a示出了在液态氧吹扫以及再生和xe回收步骤中吸附床的出口处的ch4解吸的浓度与时间的曲线。
图4b绘制了在液态氧吹扫以及再生和xe回收步骤中吸附床的出口处的kr解吸的浓度与时间的曲线。
图4c绘制了在液态氧吹扫以及再生和xe回收步骤中吸附床的出口处的xe解吸的浓度与时间的曲线。
图5示出了在比较例1的气相提取步骤中吸附床的出口处的浓度曲线,比较例1示出了未满足xe出口浓度标准(顶部),并且在床加热期间超过进料浓度的显著量的甲烷与氙一起被解吸(底部)。
图6示出了在室温气提步骤、低温气提步骤(130k)、吹扫和xe回收以及床再生期间吸附床的出口处的浓度曲线。
图7示意性地示出了分别在步骤1a、步骤1b和步骤2中用液态进料和吹扫进行1-床吸附方法的kr和xe回收方法步骤。
图8示出了在进料步骤中吸附床的出口处的kr和ch4的浓度与时间的曲线。
具体实施方式
本发明涉及一种用于从低温氧流体流中回收氙的吸附方法,该方法可在氧气可在再生期间安全地用作吹扫气体的条件下操作,并且其中该方法可在有利于吸附氙以及其它组分的压力和温度条件下操作。
在一个实施方案中,本发明涉及一种从低温氧流体流中回收氙的吸附方法,其中:
1.使具有入口和出口的吸附床在低温下与包含氧、氙和至少一种其它可吸附组分(诸如氪和/或甲烷)的流体进料流接触。将吸附床保持在进料上,直到床的出口处的氙浓度大于或等于床的入口处的氙浓度的70%;在另一个实施方案中,大于或等于床的入口处的氙浓度的80%,并且在又一个实施方案中,大于或等于床的入口处的氙浓度的90%。将吸附床保持在进料上直到床的出口处的氙浓度水平高于某一浓度水平的原因是为了实现对床中所有不需要的组分、尤其是烃的深度排斥。更具体地讲,由于氙在吸附剂上的吸附比低温稀有气体流中通常存在的轻质烃更强,因此将床保持在进料上直到床的出口处的氙浓度大于或等于床的入口处的氙浓度的70%,迫使吸附在吸附剂上的任何烃被氙取代,从而实现对床中的烃的深度排斥。这类烃的示例包括但不限于甲烷、乙烷、丙烷、乙烯及其组合。出于明显的安全原因,富集具有大于标称浓度的c1-c3烃的基于氧气的流体进料流是不利的,如果未满足出口浓度标准,则可在吸附床中发生这种情况。
2.此时,结束进料步骤并用吹扫气体吹扫吸附床,以主要从床的非选择性空隙空间中去除烃和其它可吸附组分。吹扫气体通常包含氧气、氮气、氩气和/或其混合物。在一个实施方案中,吹扫气体基本上不含氙气和/或其它可吸附组分。
3.将吸附床的温度升高到足以解吸氙产物的温度,可按原样收集该氙产物和/或对其进行进一步加工以进一步提高其纯度。
4.用低温流体将吸附床冷却至低温。在一个实施方案中,低温流体基本上不含氙气和其它可吸附组分。在另一个实施方案中,低温流体包含氧气和/或液体。
5.以循环方式重复步骤1-4。
本发明的方法可利用一个或多个吸附床进行操作,并且可与其它吸附剂和/或方法一起使用,这可有助于简化进入氙回收吸附材料(诸如凝胶阱或保护床)的进料流。有用的吸附剂包括与银进行离子交换的沸石,其具有小于3的低二氧化硅与氧化铝比率以及足以吸附氙的孔。可用于本发明的方法的吸附剂的非限制性示例包括x型或lsx型的银离子交换沸石(其中lsx表示x型沸石的低二氧化硅变体),其中离子交换水平以当量计为至少80%ag;在另一个实施方案中,以当量计为至少90%ag。
本发明的吸附方法主要从流体流(液相或气相)中回收氙,而不产生对烃的富集高于和超出其在进料流中的浓度的产物。事实上,当以优选方式操作时,氙产物流中的烃含量从含有高达2000ppm的进料降低至小于或等于50ppm,优选小于或等于1ppm。当吸附剂远离饱和,即没有实现完全穿透时,吸附剂将吸附下一个最优物质,我们的数据显示这些物质包含烃,包括甲烷。随着更多的氙被引入床中,甲烷被置换并从吸附剂中释放出来,从而可将其吹扫出去。将吸附剂操作至完全穿透确保了吸附剂中没有剩余容量用于烃诸如甲烷,因此相同和其它烃的唯一来源将是吸附剂颗粒与吸附床中其它地方之间的空隙空间。通过吹扫该空隙空间,甲烷/其它烃被有效地去除,从而减少了氙产物的收集步骤中的潜在危险。
由于通过本发明的吸附方法实现了对烃的深度排斥,因此避免了复杂的解吸方案或使用惰性吹扫气体诸如氮气和/或添加其它单元操作诸如催化烃去除。此外,我们能够处理含低温液态氧的进料流避免了对蒸发的需要,当存在烃时,蒸发可能由于烃在蒸发期间在氧流体中的浓度而引起安全问题。
本发明的一个经济优点源在于方法的简单性,因此需要较少的资本设备。例如,能够处理液体进料流使得不需要蒸发器将液体转化为气体。能够使用氧气吹扫气体意味着较少的接头和/或补充管线,例如不需要按照golden等人的技术添加氮气管线。产物中不含烃也简化了下游加工,这意味着不需要额外的资本设备,诸如催化式焚烧炉和下游预净化器来去除燃烧产物。
在另一个实施方案中,本发明涉及一种将产物中的烃深度排斥至50ppm或更低并从包含至少ppm水平的氙和烃的低温氧流体进料流中回收浓度≥1%的氙的吸附方法。其它物质(包括二氧化碳、一氧化二氮和氪)也可存在于进料流中。合适的流体流的示例包括:
·xe1ppm-200ppm,在另一个实施方案中为20ppm-180ppm,在另一个实施方案中为50ppm-150ppm
·kr500ppm-2000ppm
·烃(如甲烷)500ppm-2000ppm
·n2o0ppm-100ppm
·co20ppm-100ppm
·ar0ppm-1200ppm
·ppm量的其它大气气体,包括n2,并且
·余量为o2
上述富氧流优选处于≤120k的低温下,在另一个实施方案中处于≤90k的低温下,并且被加压至至少10psig。如果富氧流含有比xe更强吸附的组分,诸如co2和n2o,则优选基本上去除这些物质,然后通过使用主吸附容器内的凝胶阱、保护床和/或吸附层来使进料流与主吸附床接触,该主吸附容器容纳有用于xe回收的吸附剂。用于除去这些强吸附物质的合适吸附剂是硅胶。在一个实施方案中,将硅胶成型为与填充床吸附方法相容的形式,诸如颗粒状或珠粒状。在另一个实施方案中,成型材料的平均粒度为至少0.5mm且不超过约5mm。在一个实施方案中,用于xe回收的吸附剂为ag交换的沸石,在另一个实施方案中为agx沸石,其中ag交换度以当量计为至少80%,在另一个实施方案中为至少90%。也可有利地将agx沸石部署为成型颗粒,其中可使用珠粒、挤出物或颗粒材料。平均粒度也有利地为至少0.5mm且不超过约5mm。
在另一个实施方案中,本发明的方法可用两个或更多个吸附容器来实践。吸附容器可以是任何已知类型,包括垂直流动容器、水平流动容器、侧向流动容器或径向流动容器。当用两个或更多个容器来实践本发明的方法时,可改变床的操作阶段,使得当第二床离线时第一床的在线时段开始,从而使得存在最小的产出波动。或者,可对循环分阶段,以使床的在线时段之间存在重叠。
吸附方法描述:氙回收吸附床
步骤1:提取步骤
将含有对xe具有选择性的吸附剂的吸附床用低温氧流体预冷至≤120k,在另一个实施方案中预冷却至≤90k。在≤120k下,在另一个实施方案中在≤90k下并且在约10psig的压力下,提供在氧基质中含有至少ppm水平的xe和烃的进料流。随着进料流继续流动,对xe有选择性的吸附剂被xe逐渐饱和。有意地继续进料步骤,直到床的出口处的xe浓度为床的入口处的xe浓度的至少70%,在另一个实施方案中为至少80%,在另一个实施方案中为至少90%,并且在又一个实施方案中为至少95%。此时,结束进料流。
步骤2:吹扫步骤
一旦达到结束步骤1的标准,即床的出口处的xe浓度为床的入口处的xe浓度的至少70%,在另一个实施方案中为至少80%,在另一个实施方案中为至少90%,并且在又一个实施方案中为至少95%,便用选自氧气、氮气、氩气或其混合物的合适吹扫气体在≤120k下吹扫床,以从吸附床中存在的非选择性空隙中除去烃和xe。应当继续该吹扫步骤,直到吸附床的出口处的烃水平为≤50ppm;在另一个实施方案中为≤10ppm,并且在又一个实施方案中为≤1ppm。
步骤3:再生和xe回收
此时,可将吸附床的温度从低温升高到至少250k且高达450k,以便回收浓度≥1%且含有最多50ppm的烃(以甲烷当量测量)的xe产物。通过使用环境温度或更热的吹扫气体可升高温度,并且可通过所使用的该环境温度或更热温度的吹扫气体的量来控制xe产物的纯度。
步骤4:床冷却
在吸附床的温度达到至少250k并且已经回收xe产物后,将床再次冷却至低温。这是通过停止环境温度或更暖温度的吹扫气体的流动并使吸附床与低温流体接触以将床冷却至≤120k、优选冷却至≤90k来实现的。
步骤1-4可循环操作。也可以使用一个或多个吸附床来操作该吸附方法。如果使用两个吸附床,则有利的是操作以下方法:其中按顺序操作这两个吸附床,使得当床1处于步骤1时,床2正在进行步骤2、3和4。特别有利的是,在床1记录任何xe穿透之前,操作2床方法以使床2在步骤2、3和4之后再生。通过这种方式,对于步骤1的一部分,可串联操作床1和床2以使得床1的出口xe浓度为入口浓度的至少70%,在另一个实施方案中为至少80%,在另一个实施方案中为至少90%,并且在又一个实施方案中为至少95%,同时当这些床在进料步骤的一部分中串联连接时,能够以可接受的回收率增加总xe产物纯度。
本发明的一个实施方案在步骤2和步骤3中使用氧气作为吹扫气体,在步骤4中使用低温氧气和/或液态氧作为低温流体。该实施方案在图1中示意性地示出。从减少整个方法所需的接头数量的观点来看,使用环境温度或更高温度的氧气作为吹扫并使用液态氧作为冷却流体是有利的。在步骤3中使用温度高达450k的高温吹扫气体能够减少使床再生所需的吹扫量,这可有助于提高xe产物的纯度,并且允许在更少的时间内完成再生步骤。根据需要,可使用小型装饰加热器来将吹扫气体的温度升高到环境以上的温度。
现在将通过以下非限制性实施例来说明本发明。
实施例
提供了总共四个实施例以减少本发明的关键特征来进行实践。这些实施例示出了从同时具有气相和液相进料流的不同进料流中回收氙,并且在一个实施方案中,示出了可如何改进基本吸附方法以便除了回收氙之外还回收一些氪。
·实施例1:从较低进料浓度的液体中回收xe
·实施例2:从较高进料浓度的液体中回收xe
·实施例3:从较高进料浓度的气体中回收xe
·实施例4:从进料液体中回收kr和xe
·比较例:不满足xe穿透标准的影响。
如实施例所示,可根据需要在提取步骤(步骤1)中使用气相或液相进料来实践本发明。实施例1和实施例2为液相实验,而实施例3为气相实验。此外,可调整本发明的方法以允许除了回收氙之外还回收一些氪(实施例4)。
实施例1:从含50ppm的xe、500ppm的ch4、余量为o2的进料液体中回收xe。
将5.568g的平均粒度为0.6mm的aglsx珠粒(以当量计,99%用ag交换,余量为na)填充到不锈钢吸附床(0.62英寸内径×3.5英寸高度)中,该吸附床配备有入口和出口以及放置在吸附床的中点附近用于测量温度的热电偶。在干燥氮气下,将aglsx吸附剂在350℃下活化4小时,以将残余水分含量降低至≤0.5重量%,如通过卡尔费休滴定法测量。将这种现在活化的吸附床置于来自oxfordinstruments的液氮冷却低温恒温器内,其中可将吸附床的温度在77k-300k的整个温度范围内控制在±1℃内。吸附床的入口连接到歧管,使得含50ppm的xe、493ppm的ch4、余量为o2的进料能够流过吸附床或者使得用作吹扫流体的uhp级o2能够流过吸附床。使用放置在出口管线上和吸附床后的背压调节器来控制该测试装置中的压力。除非另外指明,否则提取步骤期间的压力为100psig,并且吹扫步骤和温度摆动床再生期间的压力为50psig。使用来自pffeifervacuumag的具有200原子质量单位的omnistar残余气体分析仪(rga)来测量离开床的气体的组成,每个数据点的时间分辨率为约0.1分钟。该测试装置由阀门完成,使得进料和/或吹扫流体能够在并流或逆流流动路径中通过吸附床。还包括旁通回路,以使得能够绕过床,从而使得能够在任何时间测量进料组成,以及有利于rga的校准。使用该装置来测量所有非o2组分(包括xe和ch4)的穿透曲线,以及所有非o2组分的解吸浓度曲线。解吸曲线的目的是检测在xe回收步骤3中去除的ch4是否满足该方法的需求(例如,ch4浓度≤50ppm)。
用uhpo2吹扫吸附床以去除任何大气污染物,然后将其冷却至86k,并使用uhpo2以1.5slpm的气体当量流速将其加压至100psig。当床处于该温度时,使含有xe和ch4的进料混合物在相同的压力、温度和流动条件下与吸附床接触。5小时后,rga检测到ch4的初始穿透。在约6.5小时的进料时间后,ch4浓度达到入口进料浓度。此时,没有发生xe穿透。继续进料流并在100.5小时后测量到xe的初始穿透。在145小时的进料时间后,实现了xe穿透到50ppm的入口浓度(c/c0=1.0)。此时,结束进料流并开始在0.5slpm下用uhpo2流清洁床中的非选择性空隙以及xe和ch4进料组分的管道。同时,将床温度升高至130k。一旦ch4浓度<100ppm,便将吸附床以2k/分钟的速率从130k加热至300k,将吸附床压力降低至50psig,并在o2吹扫下将xe从吸附床上解吸。在实际xe解吸开始时,ch4浓度为1ppm并在1.2小时后降至0ppm。温度达到约300k后6小时,xe解吸基本上完成。进料和解吸步骤的浓度曲线在图2中示出。
实施例2:从含150ppm的xe、1500ppm的kr、1540ppm的ch4、50ppm的n2o、余量为o2的进料液体中回收xe。
用1.83g的平均粒度为1.0mm的aglsx(以当量计,99%用ag交换,余量为na)填充实施例1的吸附床,其仅部分填充该床。用平均粒度也为1.0mm的基本上不吸附的玻璃珠填充床中的额外空间。该变化主要是为了减少实验时间,在实施例1中所述的测试中(其中使用更多的aglsx吸附剂),实验时间超过150小时。如实施例1中所述,在使用前活化aglsx吸附剂。由于进料气体中存在n2o,将含有3.54g的40级gracedavison硅胶的凝胶阱放置在aglsx吸附床之前。在使用之前,将硅胶在200℃下活化4小时以在使用前调理该材料。凝胶阱的目的是尽可能多地去除n2o,以便使更昂贵的aglsx吸附剂能够如实施例1中所示那样起作用,从而用于xe捕获和回收。在环境温度下用uhpo2吹扫系统,以从吸附床、凝胶阱以及相关组件和管道中去除任何大气污染物。使用背压调节器将压力设置为100psig。然后使用液体n2将凝胶阱冷却至81.9k的平均温度。类似地冷却低温恒温器内的吸附床(如实施例1中所述),这次仍然在uhpo2流下冷却至85.6k的平均温度。一旦凝胶阱和吸附床温度稳定,便将进料从uhpo2切换为具有以下组成的含有xe的混合物:150ppm的xe、1502ppm的kr、1540ppm的ch4、50ppm的n2o、余量为o2。吸附步骤的流速为0.5slpm。到c/c0=1的穿透时间在表1中给出。稀有气体和污染物组分的浓度与时间曲线在图3中给出。从这些数据可以清楚地看出,首先发生kr穿透,然后是ch4,这两种物质在不到5小时内穿透。相比之下,xe花了超过52个小时才完全穿透。在该吸附步骤中,在出口处未检测到n2o。
表1:稀有气体和杂质的穿透时间。
接下来,结束含有稀有气体的混合物的进料,并且以0.5slpm开始用uhpo2吹扫并将系统压力降低至50psig,以从空隙和其它非选择性空间中吹扫吸附床和相关管道中任何痕量的含稀有气体的混合物。在吹扫步骤中,将吸附床温度逐渐升高至130k。吹扫持续时间为60分钟,然后将吸附床的温度一度逐渐升高至室温以解吸吸附的xe。在解吸步骤中,将uhpo2吹扫流降低至0.1slpm。当吸附床温度达到大约250k时,大部分xe在约2小时后解吸(参见图4a至图4c)。
实施例1和实施例2均示出了根据本文公开的方法步骤并且通过使用优选的agx吸附剂进行氙回收可获得的结果。在这两个实施例中,进料组成在氙浓度以及杂质的量和类型方面都不同。尽管存在这些进料组成差异,但是测试数据清楚地表明agx吸附剂在液相提取步骤(步骤1)中优先吸附氙组分。用氧气进行吹扫(步骤2)表明,可在步骤1结束时从床和管道中的空隙空间中容易地清除进料组分。如步骤3所述的使床变暖导致将氙从低ppm富集到百分比水平来进行回收,并且其中在两种情况下产物中的烃水平都为≤1ppm。在这两个实验中首先进行最后的冷却步骤(步骤4),以便将去除强吸附的污染物(诸如,co2和n2o)所需的吸附床以及任何凝胶阱或保护床降至低温,然后开始提取步骤(步骤1)。
比较例1:从含49.7ppm的xe、500ppm的kr、508ppm的ch4、49.2ppm的n2o、余量为o2的进料气体中回收xe,其中不满足提取步骤的xe穿透标准。
用1.85g的平均粒度为1.0mm的aglsx(以当量计,99%用ag交换,余量为na)填充实施例1的吸附床,其仅部分填充该床。用平均粒度也为1.0mm的基本上不吸附的玻璃珠填充床中的额外空间。如实施例1中所述,在使用前活化aglsx吸附剂。由于进料气体中存在n2o,将含有2.85g的40级gracedavison硅胶的凝胶阱放置在aglsx吸附床之前。如实施例2中所述,在使用前活化硅胶。如实施例2中所讨论的,凝胶阱的目的是尽可能多地去除n2o。在环境温度下用uhpo2吹扫系统,以从吸附床、凝胶阱以及相关组件和管道中去除任何大气污染物。然后使用液体n2将凝胶阱冷却至81.9k的平均温度。类似地冷却低温恒温器内的吸附床(如实施例1中所述),这次仍然在uhpo2流下冷却至130k的平均温度,并且使用背压调节器将压力设置为60psig,将其放置在吸附床之后。一旦凝胶阱和吸附床温度稳定,便将进料从uhpo2切换为具有以下组成的含有xe的混合物:49.7ppm的xe、500ppm的kr、508ppm的ch4、49.2ppm的n2o、余量为o2。吸附步骤的流速为1.0slpm。继续进料步骤5小时,此时没有发生xe穿透(参见图5)。相比之下,此时,kr和ch4都已完全穿透。为了使床再生并回收xe产物,使用以下顺序的步骤:首先,在130k和60psig下用uhpo2吹扫吸附床和管道3分钟,以至少清洁吸附床和管道的空隙中的进料污染物。在该高压吹扫之后,使用背压调节器将吸附床压力降低至6psig,并且在130k下继续用uhpo2吹扫另外20分钟。在该低压吹扫之后,将流速降低至0.1slpm并用n2吹扫吸附床4分钟,以试图进一步从吸附床中去除可能更强保持的污染物,因此使用更极性的n2代替o2用作吹扫气体更容易去除这些污染物。在该短暂的n2吹扫之后,启动并流o2吹扫大约90分钟以吹扫吸附床和不含n2的管线,不含n2的管线会干扰rga分析仪上的ch4信号。此时,将气体切换回o2,其中流动方向是逆流的,并且将床的温度升高到300k。在吸附床升温期间,将流速设置为0.5slpm。如图5所示,在这些条件下,xe达到峰纯度2075ppm,并且ch4峰纯度也非常高,为3180ppm。
在该比较例中,没有遵循关于需要使xe穿透至进料浓度的至少90%并且优选其在进料中的浓度的至少95%的教导。因此,回收的xe的纯度更低,并且更显著的是,xe产物中的ch4浓度超过其在进料气体中的浓度(3180ppm对508ppm)。
实施例3:从含150ppm的xe、1502ppm的kr、1540ppm的ch4、50ppm的n2o、余量为o2的进料气体中回收xe。
如前所述,将1.85g的平均粒度为1.0mm的aglsx吸附剂装入吸附床中,用平均粒度为1.0mm的玻璃珠填充另外的空隙空间。如实施例1中所述,在使用前活化aglsx吸附剂。如实施例2中所述,使用2.85g的相同类型的硅胶,同样将凝胶阱用于该测试。如实施例2中所述活化硅胶。在开始测试之前,在室温下用uhpo2吹扫包括凝胶阱、吸附床和所有相关管道在内的整个测试装置,以去除任何大气污染物。使用背压调节器将压力设置为100psig。仍然在uhpo2流下将凝胶阱冷却至平均温度81.9k。有意将吸附床保持在室温下,以在环境温度条件下研究xe捕获量。一旦保护床处于该温度,便将进料切换为含有以下各项的稀有气体混合物:150ppm的xe、1502ppm的kr、1540ppm的ch4、50ppm的n2o和余量o2。将流速设置为0.35slpm。在室温进料的情况下,在约8小时内实现了xe穿透到进料浓度。接下来,使用低温恒温器将吸附床温度从室温降低至130k,并将流速增加至2.5slpm。在降低温度后,吸附床的出口处的xe浓度降低,表明在该较低但仍为气相的温度下发生了更多的xe吸附。在130k下大约8小时后,再次重新建立xe到进料浓度的穿透。此时,将进料从含稀有气体的混合物更换为uhpo2,将压力降至50psig,并将吹扫步骤的流速降至0.5slpm。吹扫步骤持续60分钟,然后将床温度从130k逐渐升高到环境温度,以进行xe回收和床再生的步骤。如图6所示,在xe回收步骤中没有检测到ch4,但是从吸附剂中释放出少量n2o,最可能的原因是使用2.5slpm的高流速,减少了总体测试时间。
本发明的方法还可适用于从含有至少kr、xe、ch4和余量为氧的气体或液体流中回收氪。图7示出了其中在步骤2和步骤3中将氧气用作吹扫气体并且将液态氧用于床冷却步骤4的一个实施方案。允许从该气体混合物中分离氪的一种方法变型依赖于agx吸附剂对该组分的亲和力与甲烷或氙相比是最小的。因此,在甲烷和/或其它烃和氙之前,首先发生不那么强保持的氪的穿透。因此,在不可接受量的甲烷和/或其它烃穿透之前,可在此期间回收一些氪。氪和氙回收的吸附方法包括以下步骤:
步骤1a:提取步骤和kr回收
对于含有对xe有选择性的吸附剂且用低温氧流体预先冷却至≤120k、优选冷却至≤90k的吸附床,同样在≤120k、优选≤90k下并且在约10psig的压力下将在氧基质中含有至少ppm水平的kr、xe和烃的进料流提供给吸附床的入口。对xe有选择性的吸附剂能够在提取步骤的早期阶段通过更强地吸附烃并使kr首先穿透吸附床而实现烃/kr分离。此时可回收kr,直到吸附床的出口处的烃含量超过10ppm。
步骤1b:提取步骤
一旦出口处的烃含量超过10ppm,便结束kr的回收,并且随着进料流的流动继续,提取步骤继续使xe选择性agx吸附剂被xe逐渐饱和。有意地继续进料步骤,直到床的出口处的xe浓度为床的入口处的xe浓度的至少90%,优选至少95%。此时,结束进料流。
步骤2:液态氧吹扫步骤
一旦达到结束步骤1b的标准,即床的出口处的xe浓度为床的入口处的xe浓度的至少90%,则应用选自氧气、氮气、氩气及其混合物的吹扫气体在≤120k下吹扫床,以从吸附床中存在的非选择性空隙中除去烃和xe。应当继续该吹扫步骤,直到吸附床的出口处的烃水平≤50ppm,优选≤10ppm,并且最优选≤1ppm。
步骤3:再生和xe回收
此时,可将吸附床的温度从低温升高到至少250k,以便回收百分比水平且含有最多50ppm的烃(以甲烷当量测量)的xe产物。通过使用环境温度或更热温度(高达450k)的氧气可升高温度,并且可通过所使用的该环境温度的吹扫气体的量来控制xe产物的纯度。
步骤4:床冷却
在吸附床的温度达到至少250k并且已经回收xe产物后,必须将床再次冷却至低温。这是通过停止环境温度或更暖温度的吹扫气体的流动并使吸附床与低温流体接触以将床冷却至≤120k、优选冷却至≤90k来实现的。
步骤1-4可循环操作。也可以使用一个或多个吸附床来操作该吸附方法。如果使用两个吸附床,则有利的是操作以下方法:其中这两个吸附床彼此异相,使得床1处于步骤1a和步骤1b,床2正在进行步骤2、步骤3和步骤4。特别有利的是,在床1记录任何xe穿透之前,操作2床方法以使床2在步骤2、步骤3和步骤4之后再生。通过这种方式,对于步骤1a和步骤1b的一部分,可串联操作床1和床2以使得床1的出口xe浓度大于入口浓度的95%,同时增加整体xe和kr回收。如图7所示的优选实施方案是在步骤2和步骤3中使用氧气作为吹扫气体,在步骤4中使用液态氧作为冷却流体。
实施例4:从含150ppm的xe、1500ppm的kr、1540ppm的ch4、50ppm的n2o、余量为o2的进料液体中回收kr。
用1.83g的平均粒度为1.0mm的aglsx(以当量计,99%用ag交换,余量为na)填充实施例1的吸附床,其仅部分填充该床。用平均粒度也为1.0mm的基本上不吸附的玻璃珠填充床中的额外空间。如实施例1中所述,在使用前活化aglsx吸附剂。由于进料气体中存在n2o,将含有3.54g的40级gracedavison硅胶的凝胶阱放置在aglsx吸附床之前。如实施例2中所述,在使用前将其活化。在环境温度下用uhpo2吹扫系统,以从吸附床、凝胶阱以及相关组件和管道中去除任何大气污染物。使用背压调节器将压力设置为100psig。然后使用液体n2将凝胶阱冷却至81.9k的平均温度。类似地冷却低温恒温器内的吸附床(如实施例1中所述),这次仍然在uhpo2流下冷却至85.6k的平均温度。一旦凝胶阱和吸附床温度稳定,便将进料从uhpo2切换为具有以下组成的含有xe的混合物:150ppm的xe、1500ppm的kr、1540ppm的ch4、50ppm的n2o、余量为o2。吸附步骤的流速为0.5slpm。kr、ch4和xe的穿透曲线在图8中给出。从该图中的数据可以清楚地看出,kr穿透发生在ch4穿透之前,使得在ch4穿透发生之前能够回收一些kr。
- 还没有人留言评论。精彩留言会获得点赞!