一种低温合成片状ZrB2超细粉体的方法与流程
本发明属于硼化物高温陶瓷制备技术领域,特别涉及一种低温合成片状zrb2超细粉体的方法。
背景技术
二硼化锆(zrb2)陶瓷作为超高温陶瓷材料,具有高熔点、高硬度、高弹性模量、高导热导电系数、化学性质稳定等优点,是火箭、超音速冲压喷射发动机、核反应堆反应器的耐火涂层等超高温工作环境最有潜力的候选材料。然而zrb2陶瓷材料的烧结性能受原始粉末特性的制约,如zrb2粉体的颗粒尺寸、微观结构、纯度等。例如目前合成的zrb2超细粉体主要为粒状、短棒状和板状,虽然能够赋予通过zrb2超细粉体烧结成的zrb2陶瓷有益的耐高温、高硬度等优点,但也导致采用这种结构的zrb2超细粉体烧结成的zrb2陶瓷的韧性低,从而限制了zrb2陶瓷的应用。
zrb2粉体的制备方法有很多,主要是利用锆和锆的氧化物进行硼化反应。按反应物状态不同可分为固相法、液相法和气相法。其中固相法包括直接合成法、碳热还原法、金属热还原法、高温自蔓延合成法、电化学合成法等。液相法包括共沉淀法、溶胶-凝胶法等。气相法包括pvd、cvd等。在各种zrb2粉体的制备方法中,碳热还原法和高温自蔓延合成法是最常用的两种方法,但是高温自蔓延合成法直接采用单质zr和b为原料,由于这两种单质材料的高纯化成本高,且一般需要mg或al点燃反应,最终生成的zrb2粉体中金属杂质如fe、mg、al等杂质都比较高,且合成成本高。而碳热还原法虽然采用纯度较高且易得的zro2、h3bo3、c为原料,但是在碳化过程中往往伴生碳化物,从而降低了zrb2粉体的纯度。而且碳热还原法制备得到的zrb2粉体往往呈条棒状,条棒尺寸为微米级,有时甚至超过10μm,这限制了它在陶瓷材料领域的应用。
技术实现要素:
本发明的目的在于,针对现有目前zrb2超细粉体合成方面存在的上述问题,提供一种低温合成片状zrb2超细粉体的方法。本发明的合成方法中烧结温度较现有技术中的液相先驱体法降低了50℃~100℃,相较于现有技术中的固相法降低了250℃~300℃,节约能源。且合成的zrb2超细粉体为片状结构。
本发明首先利用液相先驱体法合成具有长链结构的锆质先驱体;然后经过低温裂解后,通过添加过渡金属催化剂和熔盐控制zrb2超细粉体的形貌;最后,再经过碳热还原烧结得到片状zrb2超细粉体。与固相还原工艺相比,液相先驱体法反应物料在分子或原子尺度上均匀混合,更有利于低温下得到微纳米级且物相分布均匀的高纯粉体。
为了实现上述目的,本申请采用的技术方案为:
一种低温合成片状zrb2超细粉体的方法,其特征在于,所述zrb2超细粉体为片状结构;
所述zrb2超细粉体的合成步骤如下:
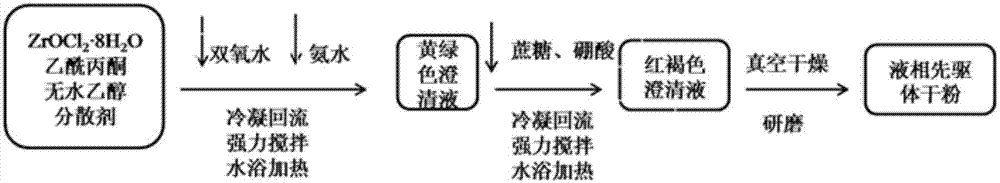
s1、锆质先驱体粉末的制备
(1)将zrocl2·8h2o、乙酰丙酮、无水乙醇、双氧水按照摩尔比1:4:40:1的比例配置成无色溶液,待用;
(2)向步骤(1)的无色溶液中再加入peg-1000,得到反应液,加入的peg-1000的重量为zrocl2·8h2o重量的2%;然后,将反应液置于92℃~97℃的恒温水浴锅中冷凝回流,且持续搅拌0.5h~1h;最后,再调节反应液ph为5~7,继续持续搅拌0.5h~1h,得到黄绿色反应液;
(3)向步骤(2)的黄绿色反应液中再加入蔗糖和硼酸,其中蔗糖与zrocl2·8h2o的摩尔比为2.5~3.5:1,硼酸与zrocl2·8h2o的摩尔为3~4:1,持续搅拌0.5h~1h,得到红褐色先驱体溶液;
(4)首先将红褐色先驱体溶液在55℃~65℃下真空干燥12h~13h;然后在105℃~115℃下真空干燥12h~13h,得到粉末;最后将得到的粉末球磨1h~2h,得到锆质先驱体粉末,待用;
s2、将s1中制备得到的锆质先驱体粉末、铁粉和氯化钠混合均匀后,置于刚玉坩埚中,并置于高温管式气氛炉中,且在惰性气体保护下煅烧,得到zrb2超细粉体;
其中所述铁粉与锆质先驱体粉末的摩尔比为0.25~0.35:1,所述氯化钠与锆质先驱体粉末的摩尔比为0.5~1.0:1;
所述惰性气体流量为0.5l/min;
所述煅烧的具体升温过程为:首先以5℃/min速率升温到800℃~900℃,并在800℃~900℃下保温0.5h;然后,继续以5℃/min速率升温到1200℃;最后,以3℃/min速率升温到1450℃~1500℃,并在1450℃~1500℃下保温3h后,自然冷却降温到室温,得到zrb2超细粉体。
进一步的,s2中所述惰性气体为氩气。
进一步的,所述步骤(2)中采用氨水和蒸馏水调节反应液ph。
进一步的,所述双氧水为质量浓度30%的双氧水。
与现有技术相比,本发明的有益效果是:
(1)在合成过程中首先利用液相先驱体法合成具有长链结构的锆质先驱体;然后经过低温裂解后,通过添加过渡金属催化剂和熔盐控制zrb2超细粉体的形貌;最后,再经过碳热还原煅烧得到zrb2超细粉体。即本申请将液相先驱体法与碳热还原法相结合,不仅降低了煅烧温度,而且避免了在碳热还原过程中碳化物的出现,进而提高了产品的纯度。
(2)采用本申请的合成方法得到的zrb2超细粉体为片状结构。
附图说明
图1是制备锆质先驱体粉末的流程图;
图2是本发明实施例1添加铁粉前后粉体的xrd图谱,其中(a)、铁粉与锆的摩尔比为0.25;(b)、铁粉与锆的摩尔比为0;
图3是本发明实施例1的方法制备得到的zrb2超细粉体的sem显微结构图。
具体实施方式
为了使本发明的技术手段、创作特征、达到目的与功效易于明白了解,下面将结合本发明的具体实施例和附图,对本发明的技术方案进行清楚、完整地描述。
本发明采用的原料zrocl2·8h2o、乙酰丙酮、无水乙醇、双氧水、氨水、peg-1000、蔗糖、铁粉和氯化钠均能够通过购买得到。采用的设备磁力搅拌器、电子恒温水浴锅、真空干燥箱、球磨机和高温管式气氛炉均属于现有技术,本领域的技术人员清楚他们的的结构和使用方法。
实施例1
一种低温合成片状zrb2超细粉体的方法,步骤如下:
s1、锆质先驱体粉末的制备
(1)将1mol即321gzrocl2·8h2o、4mol乙酰丙酮、40mol无水乙醇和1mol30%的双氧水混合后配置成无色溶液,待用;
(2)向步骤(1)的无色溶液中再加入6.42gpeg-1000,得到反应液,加入的peg-1000的重量为zrocl2·8h2o重量的2%;然后,将反应液置于95℃的恒温水浴锅中冷凝回流,且持续搅拌0.5h;最后,再通过蒸馏水和氨水调节反应液ph为5~7,继续持续搅拌0.5h,得到黄绿色反应液;
(3)向步骤(2)的黄绿色反应液中再加入3mol即1026.9g蔗糖和3mol即185.4g硼酸,其中蔗糖与zrocl2·8h2o的摩尔比为3,硼酸与zrocl2·8h2o的摩尔比3,持续搅拌0.5h,得到红褐色先驱体溶液;
(4)首先将红褐色先驱体溶液在60℃下真空干燥12h;然后在110℃下真空干燥12h,得到粉末;最后将得到的粉末球磨1h,得到锆质先驱体粉末,待用;
s2、将s1中制备得到的所有的锆质先驱体粉末、0.25mol即14g铁粉和0.5mol即29g氯化钠混合均匀后,置于刚玉坩埚中,并置于高温管式气氛炉中惰性气体保护烧结,得到zrb2超细粉体;
其中铁粉与zrocl2·8h2o的摩尔比为0.25,氯化钠与zrocl2·8h2o的摩尔比为0.5;
惰性气体为氩气,且流量为0.5l/min;
烧结的具体升温过程为:首先以5℃/min速率升温到900℃,并在900℃下保温0.5h;然后,继续以5℃/min速率升温到1200℃;最后,以3℃/min速率升温到1450℃,并在1450℃下保温3h后,自然冷却降温到室温,得到zrb2超细粉体。
实施例2
一种低温合成片状zrb2超细粉体的方法,步骤如下:
s1、锆质先驱体粉末的制备
(1)将2mol即642gzrocl2·8h2o、8mol乙酰丙酮、80mol无水乙醇和2mol双氧水混合后配置成无色溶液,待用;
(2)向步骤(1)的无色溶液中再加入12.84gpeg-1000,得到反应液,加入的peg-1000的重量为zrocl2·8h2o重量的2%;然后,将反应液置于92℃的恒温水浴锅中冷凝回流,且持续搅拌0.5h;最后,再通过蒸馏水和氨水调节反应液ph为5~7,继续持续搅拌0.5h,得到黄绿色反应液;
(3)向步骤(2)的黄绿色反应液中再加入5mol即1711.5g蔗糖和7mol即432.6g硼酸,其中蔗糖与zrocl2·8h2o的摩尔比为2.5,硼酸与zrocl2·8h2o的摩尔比3.5,持续搅拌1h,得到红褐色先驱体溶液;
(4)首先将红褐色先驱体溶液在55℃下真空干燥12h;然后在105℃下真空干燥12h,得到粉末;最后将得到的粉末球磨2h,得到锆质先驱体粉末,待用;
s2、将s1中制备得到的所有的锆质先驱体粉末、0.25mol即28g铁粉和1mol即58g氯化钠混合均匀后,置于刚玉坩埚中,并置于高温管式气氛炉中惰性气体保护烧结,得到zrb2超细粉体;
其中铁粉与zrocl2·8h2o的摩尔比为0.25,氯化钠与zrocl2·8h2o的摩尔比为0.5;
惰性气体为氩气,且流量为0.5l/min;
烧结的具体升温过程为:首先以5℃/min速率升温到800℃,并在800℃下保温0.5h;然后,继续以5℃/min速率升温到1200℃;最后,以3℃/min速率升温到1450℃,并在1450℃下保温3h后,自然冷却降温到室温,得到zrb2超细粉体。
实施例3
一种低温合成片状zrb2超细粉体的方法,步骤如下:
s1、锆质先驱体粉末的制备
(1)将1mol即321gzrocl2·8h2o、4mol乙酰丙酮、40mol无水乙醇和1mol双氧水混合后配置成无色溶液,待用;
(2)向步骤(1)的无色溶液中再加入6.42gpeg-1000,得到反应液,加入的peg-1000的重量为zrocl2·8h2o重量的2%;然后,将反应液置于97℃的恒温水浴锅中冷凝回流,且持续搅拌1h;最后,再通过蒸馏水和氨水调节反应液ph为5~7,继续持续搅拌1h,得到黄绿色反应液;
(3)向步骤(2)的黄绿色反应液中再加入3.5mol即1198g蔗糖和4mol即247.2g硼酸,其中蔗糖与zrocl2·8h2o的摩尔比为3.5,硼酸与zrocl2·8h2o的摩尔比4,持续搅拌1h,得到红褐色先驱体溶液;
(4)首先将红褐色先驱体溶液在65℃下真空干燥13h;然后在115℃下真空干燥13h,得到粉末;最后将得到的粉末球磨2h,得到锆质先驱体粉末,待用;
s2、将s1中制备得到的所有的锆质先驱体粉末、0.35mol即19.6g铁粉和1mol即58g氯化钠混合均匀后,置于刚玉坩埚中,并置于高温管式气氛炉中惰性气体保护烧结,得到zrb2超细粉体;
其中铁粉与zrocl2·8h2o的摩尔比为0.35,氯化钠与zrocl2·8h2o的摩尔比为1.0;
惰性气体为氩气,且流量为0.5l/min;
烧结的具体升温过程为:首先以5℃/min速率升温到900℃,并在900℃下保温0.5h;然后,继续以5℃/min速率升温到1200℃;最后,以3℃/min速率升温到1500℃,并在1500℃下保温3h后,自然冷却降温到室温,得到zrb2超细粉体。
对比例1
对比例1与实施例1的操作步骤完全相同,不同之处仅仅是对比例1中没有添加铁粉。
对比例2
对比例2与实施例1的操作步骤完全相同,不同之处仅仅是对比例2中没有添加氯化钠。
我们将本申请的合成方法与现有合成方法相比较。目前现有的通过液相先驱体法或溶胶凝胶法制备的zrb2超细粉体的结构为粒状或短棒状,且煅烧温度为1500℃~1550℃;现有的通过固相法合成zrb2超细粉体时,煅烧温度为1700℃~1750℃,显然本申请的煅烧温度相较于现有技术中的液相先驱体法降低了50℃~100℃,相较于现有技术中的固相法降低了250℃~300℃,节约能源。
此外,为了验证通过本申请的方法合成的zrb2超细粉体的结构以及纯度,我们分别以实施例1、对比例1和对比例2制备得到的zrb2超细粉体为例进行检测分析,结果如图2和图3。
图2是本发明实施例1添加铁粉前后粉体的xrd图谱:(a)、铁粉与锆质先驱体粉末的摩尔比为0.25;(b)、铁粉与锆质先驱体粉末的摩尔比为0。将(a)和(b)进行对比发现,实施例1制备的到的zrb2超细粉体中不含有氧化锆的特征峰和碳化锆的特征峰,而对比例1制备得到zrb2超细粉体中含有杂质碳化锆,说明通过本申请的合成方法制备得到的zrb2超细粉体的纯度很高,几乎不含杂质。
虽然对比例2也能够制备得到zrb2超细粉体,但是纯度很低。
图3是本发明实施例1的方法制备得到的zrb2超细粉体的sem显微结构图;通过图3能够发现实施例1制备得到的zrb2超细粉体为片状结构,厚度为0.2μm~0.5μm,径厚比16~26。
综上所述,本申请得到的zrb2超细粉体为片状结构,且合成过程中温度相较于现有技术较低,合成的产品几乎不含杂质。
以上公开的仅为本发明的较佳实施例,但是,本发明实施例并非局限于此,任何本领域的技术人员能思之的变化都应落入本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!