制备具有CHA骨架类型的沸石材料的快速间歇方法与流程
本发明涉及一种制备具有cha骨架类型的沸石材料的超快间歇方法,其中使用含环烷基铵的结构导向剂使沸石材料结晶。
分子筛由国际沸石协会结构委员会根据iupac委员会关于沸石命名的规则进行分类。根据该分类,结构已经确定的骨架类型沸石和其它结晶微孔分子筛被赋予三个字母的代码,并描述于atlasofzeoliteframeworktypes,第5版,elsevier,英国伦敦(2001)。
在所述沸石材料中,菱沸石是一个充分研究的实例,其是具有cha骨架结构的沸石材料类别的典型代表。属于具有cha型骨架结构的分子筛类别的沸石材料被用于各种应用中,特别地用作宽范围的反应中的多相催化剂,仅举最重要的应用中的两个,例如为甲醇至烯烃催化和氮氧化物nox的选择性催化还原。cha骨架类型的沸石材料的特征在于含有双六元环(d6r)和笼的三维8元环(8mr)孔/通道体系。具有cha型骨架结构的沸石材料,特别是具有结合的铜离子的菱沸石(cu-cha),广泛用作汽车排放物中nox级分的选择性催化还原(scr)的非均相催化剂。基于小孔开口和铜离子在cha笼中的排列,这些催化剂体系具有独特的热稳定性,其在h2o存在下耐受高于700℃的温度。
对于cha的工业生产,通常使用成本密集的1-金刚烷基三甲基氢氧化铵和其它昂贵的有机模板作为其制备的合成程序中的结构导向剂。例如,us4,544,538涉及使用1n-烷基-3-喹啉环醇、n,n,n-四烷基-1-金刚烷基铵或n,n,n-三烷基-外-氨基降冰片烷作为结构导向剂来制备ssz-13,所述ssz-13沸石材料具有cha型骨架结构。与该教导相比,wo2015/185625a提供了显著的改进,因为各发明人已经开发了间歇方法,根据该方法使用含环烷基铵的结构导向剂,使得可以省去所述含金刚烷基三甲基铵的有机模板。观察wo2015/185625a的实施例,注意到结晶时间为至少7小时,如果要获得大于90%的结晶度值,则结晶时间为至少17小时,甚至高达30小时,该时间排除了将合成混合物加热至希望的结晶温度所需的时间,根据这些实施例,结晶温度为170℃。所获得的最大结晶度为92%。
在ep3020688a1中使用含金刚烷基三甲基铵的有机模板和接近200℃的温度实现了cha型沸石的快速结晶时间。与本发明相反,专利ep3020688a1要求使用金刚烷基三甲基铵结构导向剂。金刚烷基三甲基铵结构导向剂比本发明的环烷基铵sda更具反应性,如本发明的对比实施例7所示,在85℃下陈化反应介质之后,在稀释的条件下在不进行水热处理下得到33%的结晶度。从ep3020688a1看不出对比实施例6所涉及的本发明的陈化时间和在陈化工艺开始时加入晶种的关键影响。此外,ep3020688a1在该发明的实施例6中证实,当使用金刚烷基结构导向试剂时,不需要陈化就可在加热10分钟内达到77%的结晶度值。此外,ep3020688a1也未教导对比实施例4所涉及的本发明环烷基铵结构导向剂的温度限制。因此,ep3020688a1完全没有提及开发用于本发明所涉及的更廉价和反应性更低的环烷基铵基结构导向剂的快速方法,因为其没有涉及实现本发明所需的特征的组合。
特别是对于制备沸石材料的工业规模间歇方法,通常希望结晶时间尽可能短,同时实现尽可能高的结晶度。
因此,本发明的目的是提供一种改进的制备具有cha骨架类型和包含si、al、o和h的骨架结构的沸石材料的间歇方法,其使用环烷基铵化合物作为结构导向剂且特征在于非常短的结晶时间,同时所得沸石材料的结晶度至少与wo2015/185625a的实施例中教导的那些相当,优选更高。
令人惊讶地发现,如果设计该间歇方法以包括特定的陈化步骤,则可以提供使用环烷基铵化合物作为结构导向剂的该间歇方法。
因此,本发明涉及一种制备具有cha骨架类型和包含si、al、o和h的骨架结构的沸石材料的间歇方法,包括:
(i)提供包含具有cha骨架类型和包含si、al、o和h的骨架结构的沸石材料的晶种材料;
(ii)制备包含si源、al源、(i)中提供的晶种材料、包含环烷基铵化合物的cha骨架结构导向剂和水的混合物,
其中所述环烷基铵化合物为包含阳离子r1r2r3r4n+的化合物,其中r1、r2、r3彼此独立地为具有1-6个碳原子的烷基,r4为5-8元环烷基,
其中在混合物中,水相对于以sio2计算的si源和晶种材料中所含的si的摩尔比为5:1至15:1,
其中所述混合物中,以na2o计算的钠相对于以sio2计算的si源和晶种材料中所含的si的摩尔比为0:1至0.1:1;
(iii)将(ii)中制备的混合物在其液态下加热至50-90℃的混合物温度,并将液态混合物在该温度范围下保持12-100小时;
(iv)在结晶容器中将(iii)的加热混合物加热至190-230℃的混合物温度,并在结晶容器中在自生压力下将混合物在该温度范围下保持0.5-10小时,从而获得包含悬浮在其母液中的具有cha骨架类型和包含si、al、o和h的骨架结构的沸石材料的固体材料。
关于根据(i)的提供晶种材料,已发现特别优选在使用前研磨给定的晶种材料。因此,提供晶种材料优选包括通过包括以下步骤的方法制备该晶种材料:
(i.1)提供具有cha骨架类型和包含si、al、o和h的骨架结构的沸石材料;
(i.2)制备包含(i.1)中提供的沸石材料和液体的悬浮液;
(i.3)研磨(i.2)中制备的悬浮液。
因此,本发明优选涉及一种制备具有cha骨架类型和包含si、al、o和h的骨架结构的沸石材料的间歇方法,包括:
(i)提供包含具有cha骨架类型和包含si、al、o和h的骨架结构的沸石材料的晶种材料,包括:
(i.1)提供具有cha骨架类型和包含si、al、o和h的骨架结构的沸石材料;
(i.2)制备包含(i.1)中提供的沸石材料和液体的悬浮液;
(i.3)研磨(i.2)中制备的悬浮液;
(ii)制备包含si源、al源、(i)中提供的晶种材料、包含环烷基铵化合物的cha骨架结构导向剂和水的混合物,
其中所述环烷基铵化合物为包含阳离子r1r2r3r4n+的化合物,其中r1、r2、r3彼此独立地为具有1-6个碳原子的烷基,r4为5-8元环烷基,
其中在混合物中,水相对于以sio2计算的si源和晶种材料中所含的si的摩尔比为5:1至15:1,
其中所述混合物中,以na2o计算的钠相对于以sio2计算的si源和晶种材料中所含的si的摩尔比为0:1至0.1:1;
(iii)将(ii)中制备的混合物在其液态下加热至50-90℃的混合物温度,并将液态混合物在该温度范围下保持12-100小时;
(iv)在结晶容器中将(iii)的加热混合物加热至190-230℃的混合物温度,并在结晶容器中在自生压力下将混合物在该温度范围下保持0.5-10小时,从而获得包含悬浮在其母液中的具有cha骨架类型和包含si、al、o和h的骨架结构的沸石材料的固体材料。
关于根据(i.1)的提供具有cha骨架类型和包含si、al、o和h的骨架结构的沸石材料,不存在特别的限制。通常,可设想例如其通过如在具有cha骨架类型和包含si、al、o和h的骨架结构的沸石材料中所述的方法制备。此外,可能优选的是其通过本发明的方法制备。此外,可能优选的是其通过如wo2015/185625a所述的方法制备。因此,优选(i.1)包括制备具有cha型骨架结构的沸石材料,所述骨架结构包含sio2和al2o3,其中所述方法包括以下步骤:
(a)提供包含一种或多种sio2源、一种或多种al2o3源、一种或多种含四烷基铵阳离子r1r2r3r4n+的化合物和作为结构导向剂的一种或多种含四烷基铵阳离子r5r6r7r8n+的化合物的混合物;
(b)使所得混合物结晶,从而获得具有cha型骨架结构的沸石材料;其中r1、r2、r3、r4、r5、r6和r7彼此独立地表示烷基,并且其中r8表示环烷基;
其中r1、r2、r3、r4、r5、r6和r7彼此独立地优选表示任选取代和/或任选支化的c1-c6烷基,其中r8优选表示任选杂环和/或任选取代的5-8元环烷基,其中结晶优选在溶剂热条件下进行,其中步骤(1)中提供的混合物不含任何显著量的含三甲基苄基铵的化合物,其中步骤(a)中提供的混合物进一步包含晶种。因此,还优选根据(i.1)提供的沸石材料优选为可通过或通过所述方法获得的沸石材料,其中所述沸石材料更优选表现出以下参数中的一个或多个:
-3,720-3,740cm-1的第一吸收带(b1);和1,850-1,890cm-1的第二吸收带(b2);
其中第一吸收带的最大吸光度与第二吸收带的最大吸光度之比b1:b2为0.5-1.55;
-所述沸石材料的粒度dv10为400-2,500nm;
-所述沸石材料的粒度dv50为600-3,500nm;
-所述沸石材料的粒度dv90为1,200-4,500nm;
-不含任何显著量的元素p和/或as;
-所述沸石材料的29simasnmr包含-102.0至-106.0ppm的第一峰(p'1);
和-108.0至-112.5ppm的第二峰(p'2);其中所述沸石材料的29simasnmr中的第一峰和第二峰的积分提供了0.05-0.90的积分值p'1:p'2;
-沸石骨架结构的sio2:al2o3摩尔比为4:1至200:1。
关于根据(i.1)的方法和沸石材料的任何其它优选特征,参考wo2015/185625a的相应公开内容,其内容通过引用全部并入本文。
关于根据(i.2)的制备悬浮液,优选所述制备包括将(i.1)中提供的沸石材料与液体混合,其中所述液体优选包含水,其中如果所述液体由水组成,则更优选95-100重量%,更优选99-100重量%,且其中沸石材料相对于液体的重量比优选为1:10至1:50,优选为1:20至1:40,更优选为1:25至1:35。更优选的范围为1:25至1:28或1:28至1:32或1:32至1:35。
关于根据(i.3)的研磨悬浮液,优选包括珠磨悬浮液。悬浮液研磨的时间优选为10-240分钟,更优选为20-200分钟,更优选为30-150分钟。更优选的范围为60-140分钟,更优选90-130分钟。优选使用的珠粒具有100-500微米,更优选200-400微米,更优选250-350微米的直径。珠磨机通常可在任何速率下操作,其中优选在1,000-5,000rpm,优选2,000-4,000rpm,更优选2,500-3,500rpm下操作。本文所用的术语“rpm”是指“转/分钟”。
优选地,步骤(i)可由(i.1)、(i.2)和(i.3)组成。根据本发明的该方面,由(i.3)获得的研磨浆料原样使用并用作晶种材料。
根据本发明的进一步优选的方面,可能优选的是根据(i)的提供晶种材料进一步包括,在(i.3)之后,
(i.4)将由(i.3)获得的研磨悬浮液中所含的沸石材料与液体分离。
根据该方面,优选(i.4)包括对由(i.3)获得的研磨悬浮液进行固液分离,优选包括过滤和离心中的一种或多种,从而获得分离的沸石材料,并且任选在温度为20-100℃的气体气氛中干燥分离的沸石材料从而获得干燥的晶种材料,其中所述气体气氛优选包括氧气和氮气中的一种或多种。进一步地,根据这一方面,优选所述制备晶种材料的方法由(i.1)、(i.2)、(i.3)和(i.4)组成。
因此,本发明优选涉及一种制备具有cha骨架类型和包含si、al、o和h的骨架结构的沸石材料的间歇方法,包括:
(i)提供包含具有cha骨架类型和包含si、al、o和h的骨架结构的沸石材料的晶种材料,包括:
(i.1)提供具有cha骨架类型和包含si、al、o和h的骨架结构的沸石材料;
(i.2)制备包含(i.1)中提供的沸石材料和液体的悬浮液;
(i.3)研磨(i.2)中制备的悬浮液;
(i.4)将由(i.3)获得的研磨悬浮液中所含的沸石材料与液体分离;
(ii)制备包含si源、al源、(i)中提供的晶种材料、包含环烷基铵化合物的cha骨架结构导向剂和水的混合物,
其中所述环烷基铵化合物为包含阳离子r1r2r3r4n+的化合物,其中r1、r2、r3彼此独立地为具有1-6个碳原子的烷基,r4为5-8元环烷基,
其中在混合物中,水相对于以sio2计算的si源和晶种材料中所含的si的摩尔比为5:1至15:1,
其中所述混合物中,以na2o计算的钠相对于以sio2计算的si源和晶种材料中所含的si的摩尔比为0:1至0.1:1;
(iii)将(ii)中制备的混合物在其液态下加热至50-90℃的混合物温度,并将液态混合物在该温度范围下保持12-100小时;
(iv)在结晶容器中将(iii)的加热混合物加热至190-230℃的混合物温度,并在结晶容器中在自生压力下将混合物在该温度范围下保持0.5-10小时,从而获得包含悬浮在其母液中的具有cha骨架类型和包含si、al、o和h的骨架结构的沸石材料的固体材料。
所用晶种材料的用量优选使得在(ii)中制备的混合物中,晶种材料相对于以sio2计算的si源中所含的si的重量比为0.025:1至0.15:1,优选为0.030:1至0.13:1,更优选为0.035:1至0.11:1。
因此,本发明优选涉及一种制备具有cha骨架类型和包含si、al、o和h的骨架结构的沸石材料的间歇方法,包括:
(i)提供包含具有cha骨架类型和包含si、al、o和h的骨架结构的沸石材料的晶种材料,包括:
(i.1)提供具有cha骨架类型和包含si、al、o和h的骨架结构的沸石材料;
(i.2)制备包含(i.1)中提供的沸石材料和液体的悬浮液;
(i.3)研磨(i.2)中制备的悬浮液;
(i.4)优选将由(i.3)获得的研磨悬浮液中所含的沸石材料与液体分离;
(ii)制备包含si源、al源、(i)中提供的晶种材料、包含环烷基铵化合物的cha骨架结构导向剂和水的混合物,
其中所述环烷基铵化合物为包含阳离子r1r2r3r4n+的化合物,其中r1、r2、r3彼此独立地为具有1-6个碳原子的烷基,r4为5-8元环烷基,
其中在混合物中,水相对于以sio2计算的si源和晶种材料中所含的si的摩尔比为5:1至15:1,
其中所述混合物中,以na2o计算的钠相对于以sio2计算的si源和晶种材料中所含的si的摩尔比为0:1至0.1:1;
其中在所述混合物中,晶种材料相对于以sio2计算的si源中所含的si的重量比为0.035:1至0.11:1;
(iii)将(ii)中制备的混合物在其液态下加热至50-90℃的混合物温度,并将液态混合物在该温度下保持5-100小时;
(iv)在结晶容器中将(iii)的加热混合物加热至190-230℃的混合物温度,并在结晶容器中在自生压力下将混合物在该温度范围下保持0.5-10小时,从而获得包含悬浮在其母液中的具有cha骨架类型和包含si、al、o和h的骨架结构的沸石材料的固体材料。
关于晶种材料的量,根据第一方面,优选在(ii)中制备的混合物中,晶种材料相对于以sio2计算的si源中所含的si的重量比为0.025:1至0.060:1,优选为0.030:1至0.055:1,更优选为0.035:1至0.045:1。此外,根据第二方面,优选在(ii)中制备的混合物中,晶种材料相对于以sio2计算的si源中所含的si的重量比为0.085:1至0.15:1,优选为0.090:1至0.13:1,更优选为0.095:1至0.11:1。
关于根据(ii)的cha骨架结构导向剂,优选r1、r2、r3彼此独立地为具有1-5个碳原子,优选1-4个碳原子,更优选1-3个碳原子,更优选1或2个碳原子,更优选1个碳原子的烷基,且其中r4为5-7元环烷基,优选5或6元环烷基,更优选6元环烷基。更优选地,r1、r2、r3为甲基,r4为环己基。优选地,根据(ii)的cha骨架结构导向剂中所含的环烷基铵化合物包括,优选是铵盐,优选卤化物、硫酸盐、硝酸盐、乙酸盐和氢氧化物中的一种或多种,更优选氯化物、溴化物、硫酸盐、硝酸盐、乙酸盐和氢氧化物中的一种或多种,更优选氯化物、溴化物和氢氧化物中的一种或多种,更优选氢氧化物。更优选地,根据(ii)的cha骨架结构导向剂中所含的环烷基铵化合物包括,优选是n,n,n-三甲基环己基氢氧化铵。
关于cha骨架结构导向剂,并且根据本发明的第一方面,可能优选的是99-100mol%,更优选99.5-100mol%,更优选99.9-100mol%的cha骨架结构导向剂由n,n,n-三甲基环己基氢氧化铵组成,其中更优选cha骨架结构导向剂不含n,n,n-三甲基-1-金刚烷基氢氧化铵,优选不含含n,n,n-三甲基-1-金刚烷基铵的化合物,更优选不含含金刚烷基铵的化合物。
关于cha骨架结构导向剂,且根据本发明的第一方面,优选根据(ii)的cha骨架结构导向剂进一步包含含阳离子r5r6r7r8n+的四烷基铵化合物,其中r5、r6、r7、r8彼此独立地为任选取代的烷基,其具有1-4个碳原子,优选1-3个碳原子,更优选1或2个碳原子,其中更优选r5、r6、r7、r8为甲基,其中取代基优选为氯和羟基中的一种或多种,更优选羟基。因此,在基团之一被取代的情况下,可能优选的是,所述四烷基铵化合物包括,优选是2-羟乙基三甲基铵化合物。优选地,根据(ii)的cha骨架结构导向剂中所含的四烷基铵化合物包括,优选是铵盐,优选为卤化物、硫酸盐、硝酸盐、乙酸盐和氢氧化物中的一种或多种,更优选为氯化物、溴化物、硫酸盐、硝酸盐、乙酸盐和氢氧化物中的一种或多种,更优选为氯化物、溴化物和氢氧化物中的一种或多种,更优选为氢氧化物。更优选地,根据(ii)的cha骨架结构导向剂中所含的四烷基铵化合物包括,优选是氢氧化四甲铵。
因此,本发明优选涉及一种制备具有cha骨架类型和包含si、al、o和h的骨架结构的沸石材料的间歇方法,包括:
(i)提供包含具有cha骨架类型和包含si、al、o和h的骨架结构的沸石材料的晶种材料,包括:
(i.1)提供具有cha骨架类型和包含si、al、o和h的骨架结构的沸石材料;
(i.2)制备包含(i.1)中提供的沸石材料和液体的悬浮液;
(i.3)研磨(i.2)中制备的悬浮液;
(i.4)优选将由(i.3)获得的研磨悬浮液中所含的沸石材料与液体分离;
(ii)制备包含si源、al源、(i)中提供的晶种材料、包含环烷基铵化合物和四烷基铵化合物的cha骨架结构导向剂和水的混合物,
其中所述环烷基铵化合物包括,优选是n,n,n-三甲基环己基氢氧化铵,且所述四烷基铵化合物包括,优选是四甲基氢氧化铵;
其中在混合物中,水相对于以sio2计算的si源和晶种材料中所含的si的摩尔比为5:1至15:1,
其中所述混合物中,以na2o计算的钠相对于以sio2计算的si源和晶种材料中所含的si的摩尔比0:1至0.1:1;
其中在所述混合物中,晶种材料相对于以sio2计算的si源中所含的si的重量比为0.035:1至0.11:1;
(iii)将(ii)中制备的混合物在其液态下加热至50-90℃的混合物温度,并将液态混合物在该温度范围下保持12-100小时;
(iv)在结晶容器中将(iii)的加热混合物加热至190-230℃的混合物温度,并在结晶容器中在自生压力下将混合物在该温度范围下保持0.5-10小时,从而获得包含悬浮在其母液中的具有cha骨架类型和包含si、al、o和h的骨架结构的沸石材料的固体材料。
优选地,在根据(ii)的cha骨架结构导向剂中,环烷基铵化合物相对于四烷基铵化合物的摩尔比为1:1-5.5:1,更优选为1.1:1至4:1,更优选为1.3:1-3:1,更优选为1.5:1至2.0:1。优选的范围例如为1.5:1至1.9:1,或1.5.1至1.8:1,或1.5:1至1.7:1,或1.5:1至1.6:1。
关于cha骨架结构导向剂,并且根据本发明的所述第二方面,优选99-100mol%,更优选99.5-100mol%,更优选99.9-100mol%的cha骨架结构导向剂由环烷基铵化合物和四烷基铵化合物组成,其中更优选cha骨架结构导向剂不含n,n,n-三甲基-1-金刚烷基氢氧化铵,优选不含含n,n,n-三甲基-1-金刚烷基铵的化合物,更优选不含含金刚烷基铵的化合物。
优选地,在(ii)中制备的混合物中,cha骨架结构导向剂相对于以sio2计算的si源和晶种材料中所含的si的摩尔比为0.20:1至0.30:1,更优选为0.22:1至0.29:1,更优选为0.25:1至0.28:1。
因此,本发明优选涉及一种制备具有cha骨架类型和包含si、al、o和h的骨架结构的沸石材料的间歇方法,包括:
(i)提供包含具有cha骨架类型和包含si、al、o和h的骨架结构的沸石材料的晶种材料,包括:
(i.1)提供具有cha骨架类型和包含si、al、o和h的骨架结构的沸石材料;
(i.2)制备包含(i.1)中提供的沸石材料和液体的悬浮液;
(i.3)研磨(i.2)中制备的悬浮液;
(i.4)优选将由(i.3)获得的研磨悬浮液中所含的沸石材料与液体分离;
(ii)制备包含si源、al源、(i)中提供的晶种材料、包含环烷基铵化合物和四烷基铵化合物的cha骨架结构导向剂和水的混合物,
其中所述环烷基铵化合物包括,优选是n,n,n-三甲基环己基氢氧化铵,所述四烷基铵化合物包括,优选是四甲基氢氧化铵;
其中在混合物中,水相对于以sio2计算的si源和晶种材料中所含的si的摩尔比为5:1至15:1,
其中所述混合物中,以na2o计算的钠相对于以sio2计算的si源和晶种材料中所含的si的摩尔比为0:1至0.1:1;
其中在所述混合物中,晶种材料相对于以sio2计算的si源中所含的si的重量比为0.035:1至0.11:1;
其中在混合物中,cha骨架结构导向剂相对于以sio2计算的si源和晶种材料中所含的si的摩尔比为0.25:1至0.28:1;
(iii)将(ii)中制备的混合物在其液态下加热至50-90℃的混合物温度,并将液态混合物在该温度范围下保持12-100小时;
(iv)在结晶容器中将(iii)的加热混合物加热至190-230℃的混合物温度,并在结晶容器中在自生压力下将混合物在该温度范围下保持0.5-10小时,从而获得包含悬浮在其母液中的具有cha骨架类型和包含si、al、o和h的骨架结构的沸石材料的固体材料。
优选地,在(ii)中制备的混合物中所含的水的量相对低。更优选地,在(ii)中制备的混合物中,水相对于si源和晶种材料中所含的以sio2计算的硅(si源中所含的si加上晶种材料中所含的si)的摩尔比为7:1至15:1,优选为9:1至12:1。
因此,本发明优选涉及一种制备具有cha骨架类型和包含si、al、o和h的骨架结构的沸石材料的间歇方法,包括:
(i)提供包含具有cha骨架类型和包含si、al、o和h的骨架结构的沸石材料的晶种材料,包括:
(i.1)提供具有cha骨架类型和包含si、al、o和h的骨架结构的沸石材料;
(i.2)制备包含(i.1)中提供的沸石材料和液体的悬浮液;
(i.3)研磨(i.2)中制备的悬浮液;
(i.4)优选将由(i.3)获得的研磨悬浮液中所含的沸石材料与液体分离;
(ii)制备包含si源、al源、(i)中提供的晶种材料、包含环烷基铵化合物和四烷基铵化合物的cha骨架结构导向剂和水的混合物,
其中所述环烷基铵化合物包括,优选是n,n,n-三甲基环己基氢氧化铵,且所述四烷基铵化合物包括,优选是四甲基氢氧化铵;
其中在混合物中,水相对于以sio2计算的si源和晶种材料中所含的si的摩尔比为5:1至15:1,
其中所述混合物中,以na2o计算的钠相对于以sio2计算的si源和晶种材料中所含的si的摩尔比为0:1至0.1:1;
其中在所述混合物中,晶种材料相对于si源中所含的以sio2计算的si的重量比0.035:1至0.11:1;
其中在所述混合物中,cha骨架结构导向剂相对于以sio2计算的si源和晶种材料中所含的si的摩尔比为0.25:1至0.28:1;
其中在混合物中,水相对于si源和晶种材料中所含的硅的摩尔比为9:1至12:1;
(iii)将(ii)中制备的混合物在其液态下加热至50-90℃的混合物温度,并将液态混合物在该温度范围下保持12-100小时;
(iv)在结晶容器中将(iii)的加热混合物加热至190-230℃的混合物温度,并在结晶容器中在自生压力下将混合物在该温度范围下保持0.5-10小时,从而获得包含悬浮在其母液中的具有cha骨架类型和包含si、al、o和h的骨架结构的沸石材料的固体材料。
关于根据(ii)的硅源,可使用每一种合适的来源。优选地,si源包括,更优选是二氧化硅和硅酸盐中的一种或多种,优选热解法二氧化硅、硅溶胶、无定形二氧化硅、硅胶、硅酸、硅酸酯、胶态二氧化硅、四烷氧基硅烷、二硅酸盐和倍半硅酸盐中的一种或多种,更优选热解法二氧化硅、二氧化硅水溶胶、硅胶、硅酸、硅酸酯、胶态二氧化硅和四烷氧基硅烷中的一种或多种,更优选热解法二氧化硅、二氧化硅水溶胶、硅胶和胶态二氧化硅中的一种或多种,更优选热解法二氧化硅、硅胶和胶态二氧化硅中的一种或多种。更优选地,si源包括,更优选是胶态二氧化硅。
特别是关于胶态二氧化硅,可使用us20170113941a中所述的活化胶态二氧化硅,即已用金属化合物在结构导向剂存在下改性的改性胶态硅溶胶,其中所述结构导向剂优选为包含环烷基铵化合物的上述结构导向剂,更优选为包含环烷基铵化合物和四烷基铵化合物的上述结构导向剂,并且其中金属可为us20170113941a中所述的金属之一,其中可能优选的是,金属不包括钠,更优选不包括碱金属。
关于根据(ii)的铝源,可使用每一种合适的来源。优选地,al源包括,更优选是氧化铝、铝酸盐和铝盐中的一种或多种,优选氧化铝和铝盐中的一种或多种,更优选氧化铝、三(c1-c5)醇盐、alo(oh)、al(oh)3、卤化铝(其中卤化铝优选为氟化铝、氯化铝和溴化铝)、硫酸铝、磷酸铝和氟硅酸铝中的一种或多种,更优选alo(oh)和al(oh)3中的一种或多种。更优选地,al源包括,更优选是al(oh)3,更优选结晶al(oh)3,更优选三水铝石。
优选地,在(ii)中制备的混合物中,以al2o3计算的al源相对于以sio2计算的si源和晶种材料中所含的si的摩尔比为0.001:1至0.5:1,更优选为0.01:1至0.1:1,优选为0.02:1至0.05:1,更优选为0.03:1至0.04:1。
因此,本发明优选涉及一种制备具有cha骨架类型和包含si、al、o和h的骨架结构的沸石材料的间歇方法,包括:
(i)提供包含具有cha骨架类型和包含si、al、o和h的骨架结构的沸石材料的晶种材料,包括:
(i.1)提供具有cha骨架类型和包含si、al、o和h的骨架结构的沸石材料;
(i.2)制备包含(i.1)中提供的沸石材料和液体的悬浮液;
(i.3)研磨(i.2)中制备的悬浮液;
(i.4)优选将由(i.3)获得的研磨悬浮液中所含的沸石材料与液体分离;
(ii)制备包含si源、al源、(i)中提供的晶种材料、包含环烷基铵化合物和四烷基铵化合物的cha骨架结构导向剂和水的混合物,
其中所述环烷基铵化合物包括,优选是n,n,n-三甲基环己基氢氧化铵,且所述四烷基铵化合物包括,优选是四甲基氢氧化铵;
其中si源包括,优选是胶态二氧化硅;
其中al源包括,优选是al(oh)3,优选结晶al(oh)3;
其中在混合物中,水相对于以sio2计算的si源和晶种材料中所含的si的摩尔比为5:1至15:1,
其中所述混合物中,以na2o计算的钠相对于以sio2计算的si源和晶种材料中所含的si的摩尔比为0:1至0.1:1;
其中在所述混合物中,晶种材料相对于以sio2计算的si源中所含的si的重量比为0.035:1至0.11:1;
其中在所述混合物中,cha骨架结构导向剂相对于以sio2计算的si源和晶种材料中所含的si的摩尔比为0.25:1至0.28:1;
其中在混合物中,水相对于si源和晶种材料中所含的硅的摩尔比为9:1至12:1;
其中在所述混合物中,以al2o3计算的al源相对于以sio2计算的si源和晶种材料中所含的si的摩尔比为0.03:1至0.04:1;
(iii)将(ii)中制备的混合物在其液态下加热至50-90℃的混合物温度,并将液态混合物在该温度范围下保持12-100小时;
(iv)在结晶容器中将(iii)的加热混合物加热至190-230℃的混合物温度,并在结晶容器中在自生压力下将混合物在该温度范围下保持0.5-10小时,从而获得包含悬浮在其母液中的具有cha骨架类型和包含si、al、o和h的骨架结构的沸石材料的固体材料。
优选地,根据本发明,95-100重量%,优选98-100重量%,更优选99-100重量%的在(ii)中制备的混合物由si源、al源、(i)中提供的晶种材料、包含环烷基铵化合物的cha骨架结构导向剂和水组成。更优选地,根据本发明,99.5-100重量%,更优选99.9-100重量%的在(ii)中制备的混合物由si源、al源、(i)中提供的晶种材料、包含环烷基铵化合物的cha骨架结构导向剂和水组成。优选地,根据本发明,在(ii)中制备的混合物中,以元素p计算的磷相对于以sio2计算的si源中和晶种材料中所含的si的摩尔比为0:1至0.001:1。
优选地,根据(ii),制备混合物包括:
(ii.1)制备包含si源、al源、包含环烷基铵化合物的cha骨架结构导向剂和水的混合物;
(ii.2)将所述晶种材料加入到(ii.1)中制备的混合物中,从而获得待实施(iii)的混合物。
关于混合物的该优选制备,进一步优选的是
(ii.1)包括:
(ii.1.1)制备包含al源和cha骨架结构导向剂的混合物,所述cha骨架结构导向剂包含环烷基铵化合物;
(ii.1.2)将(ii.1.1)中制备的混合物在10-50℃的混合物温度下搅动5-60分钟;(ii.1.3)将si源加入到由(ii.1.2)获得的混合物中;
(ii.1.4)将(ii.1.3)中制备的混合物在10-50℃的混合物温度下搅动1-30分钟,从而获得待实施(iii)的混合物。
因此,本发明优选涉及一种制备具有cha骨架类型和包含si、al、o和h的骨架结构的沸石材料的间歇方法,包括:
(i)提供包含具有cha骨架类型和包含si、al、o和h的骨架结构的沸石材料的晶种材料,包括:
(i.1)提供具有cha骨架类型和包含si、al、o和h的骨架结构的沸石材料;
(i.2)制备包含(i.1)中提供的沸石材料和液体的悬浮液;
(i.3)研磨(i.2)中制备的悬浮液;
(i.4)优选将由(i.3)获得的研磨悬浮液中所含的沸石材料与液体分离;
(ii)制备包含si源、al源、(i)中提供的晶种材料、包含环烷基铵化合物和四烷基铵化合物的cha骨架结构导向剂和水的混合物,
其中所述环烷基铵化合物包括,优选是n,n,n-三甲基环己基氢氧化铵,且所述四烷基铵化合物包括,优选是四甲基氢氧化铵;
其中si源包括,优选是胶态二氧化硅;
其中al源包括,优选是al(oh)3,优选结晶al(oh)3;
其中在混合物中,水相对于以sio2计算的si源和晶种材料中所含的si的摩尔比为5:1至15:1,
其中所述混合物中,以na2o计算的钠相对于以sio2计算的si源和晶种材料中所含的si的摩尔比为0:1至0.1:1;
其中在所述混合物中,晶种材料相对于以sio2计算的si源中所含的si的重量比为0.035:1至0.11:1;
其中在所述混合物中,cha骨架结构导向剂相对于以sio2计算的si源和晶种材料中所含的si的摩尔比为0.25:1至0.28:1;
其中在混合物中,水相对于si源和晶种材料中所含的硅的摩尔比为9:1至12:1;
其中在所述混合物中,以al2o3计算的al源相对于以sio2计算的si源和晶种材料中所含的si的摩尔比为0.03:1至0.04:1;
其中(ii)包括:
(ii.1)制备包含si源、al源、包含环烷基铵化合物的cha骨架结构导向剂和水的混合物;
(ii.2)将晶种材料加入到(ii.1)中制备的混合物中,从而获得待实施(iii)的混合物;其中(ii.2)优选包括:
(ii.1.1)制备包含al源和cha骨架结构导向剂的混合物,所述cha骨架结构导向剂包含环烷基铵化合物;
(ii.1.2)将(ii.1.1)中制备的混合物在10-50℃的混合物温度下搅动5-60分钟;
(ii.1.3)将si源加入到由(ii.1.2)获得的混合物中;
(ii.1.4)将(ii.1.3)中制备的混合物在10-50℃的混合物温度下搅动1-30分钟,从而获得待实施(iii)的混合物;
(iii)将(ii)中制备的混合物在其液态下加热至50-90℃的混合物温度,并将液态混合物在该温度范围下保持12-100小时;
(iv)在结晶容器中将(iii)的加热混合物加热至190-230℃的混合物温度,并在结晶容器中在自生压力下将混合物在该温度范围下保持0.5-10小时,从而获得包含悬浮在其母液中的具有cha骨架类型和包含si、al、o和h的骨架结构的沸石材料的固体材料。
优选地,根据(ii.1.2),将混合物在15-40℃,优选20-30℃的混合物温度下搅动,进一步优选地,根据(ii.1.2),将混合物搅动10-50分钟,更优选20-40分钟。进一步优选地,根据(ii.1.2),将混合物在0.7-2巴(绝对),更优选0.8-1.5巴(绝对),更优选0.9-1.1巴(绝对)的压力下搅动。根据(ii.1.2)的搅动优选包括机械搅动混合物,更优选搅拌混合物,优选在100-1,000rpm下,更优选在200-750rpm下,更优选在400-600rpm下。
优选地,根据(ii.1.4),将混合物在15-40℃,优选20-30℃的混合物温度下搅动。进一步优选地,根据(ii.1.4),将混合物搅动2-20分钟,更优选5-15分钟。进一步优选地,根据(ii.1.4),将混合物在0.7-2巴(绝对),更优选0.8-1.5巴(绝对),更优选0.9-1.1巴(绝对)的压力下搅动。根据(ii.1.4)的搅动优选包括机械搅动混合物,更优选搅拌混合物,优选在100-1,000rpm下,更优选在200-750rpm下,更优选在400-600rpm下。
根据本发明关于(ii.1)的优选方面,(ii.1)由(ii.1.1)、(ii.1.2)、(ii.1.3)和(ii.1.4)组成。根据本发明关于(ii)的进一步优选的方面,(ii)由(ii.1)和(ii.2)组成。
优选地,根据(iii),将(ii)制得的混合物在其液态下加热至55-80℃,更优选60-70℃的混合物温度。进一步优选地,根据(iii),将液态混合物在该温度范围下保持5-80小时,更优选20-50小时。优选的范围例如为20-30小时或25-35小时或30-40小时或35-45小时或40-50小时,优选地,根据(iii)的加热混合物,更优选加热混合物并将混合物保持在该温度下在0.7-2巴(绝对),更优选0.8-1.5巴(绝对),更优选0.9-1.1巴(绝对)的压力下进行。优选地,根据(iii),将混合物以0.2-5k/分钟,优选0.5-4k/分钟,更优选1-3k/分钟的升温速率加热到该温度。在根据(iii)的将混合物保持在该温度下期间,优选地在根据(iii)的在加热混合物并将混合物保持在该温度下期间,搅动混合物,其中所述搅动优选包括机械搅动混合物,更优选搅拌动混合物。搅拌优选在100-1,000rpm下,更优选在200-750rpm下,更优选在400-600rpm下进行。
根据本发明关于(iii)的优选方面,(iii)由加热混合物并将混合物保持在该温度下组成。
优选地,根据(iv),将(iii)的混合物加热至195-225℃,更优选200-220℃的混合物温度。优选的范围例如为200-210℃或205-215℃或210-220℃。根据(iv),优选将混合物在该温度范围下保持0.75-7.5小时,更优选1-5小时。优选的范围例如为1-3小时或2-4小时或3-5小时,优选地,根据(iv),将混合物以0.1-20k/分钟,更优选0.5-15k/分钟,更优选1-10k/分钟的升温速率下加热到该温度。优选的范围例如为1-5k/分钟或2-4k/分钟或5-10k/分钟或6-10k/分钟或7-10k/分钟或8-10k/分钟。在根据(iv)的加热混合物或将混合物保持在该温度下期间,优选在根据(iv)的在加热混合物并将混合物保持在该温度下期间,优选搅动混合物,更优选机械搅动,其中更优选搅动结晶容器。搅动结晶容器例如通过翻转结晶容器来进行。
根据(iv)的结晶可在每种合适的间歇结晶容器如高压釜或可密封的管式反应器中进行。关于可根据本发明方法和相应的工艺步骤使用的优选管式反应器,特别参考下文的实施方案62至71。优选地,管式反应器包括反应管和用于密封反应管的一个或两个密封盖。为了加热(包括根据(iv)的加热混合物并将混合物保持在该温度下)的目的,管式反应器可由加热介质从外部加热,其中加热介质可包括气态加热介质、液态加热介质或固态加热介质,优选气态加热介质或液态加热介质。也可进行蒸汽和电加热。如果加热介质是气态加热介质(优选包含在静态或连续操作的烘箱中),则在根据(iv)的加热和保持在该温度下期间,使气态加热介质静态或连续地与反应管接触。如果加热介质是液态加热介质(优选包括油,所述加热介质更优选包含在静态或连续操作的浴中),则在根据(iv)的加热和保持在该温度下期间,使液态加热介质静态或连续地与反应管接触。反应管优选由热扩散系数为3×10-6至30×10-6m2/s,更优选为5×10-6至25×10-6m2/s的材料制成。优选地,所述材料可为不锈钢。优选地,反应管的体积v/cm3与反应管的外表面积a/cm2之比v/a为0.1:1至100:1,更优选为0.2:1至60:1,更优选为0.5:1至10:1。
优选地,(iv)包括,优选由以下组成:
(iv.1)将由(iii)获得的经加热的混合物供应至反应管;
(iv.2)用所述一个或两个密封盖密封反应管;
(iv.3)加热(iii)的加热混合物并将混合物保持在该温度下,从而获得悬浮在其母液中的包含具有cha骨架类型和包含si、al、o和h的骨架结构的沸石材料的固体材料,其中在根据(iv.3)的加热期间,加热介质的温度优选为190-235℃,更优选为195-230℃,更优选为200-225℃;
(iv.4)任选地,冷却由(iv.3)获得的悬浮液;
(iv.5)打开一个或两个密封盖,并从反应管中移出由(iv.3)或(iv.4)获得的悬浮液。
优选地,本发明的方法进一步包括:
(v)将由(iv)获得的悬浮液冷却,优选冷却至15-40℃,优选20-30℃的温度。优选地,本发明的方法进一步包括:
(vi)将由(iv)或(v),优选由(v)获得的包含具有cha骨架类型和包含si、al、o和h的骨架结构的沸石材料的固体材料与其母液分离,从而获得包含具有cha骨架类型和包含si、al、o和h的骨架结构的沸石材料的固体材料。
关于根据(vi)的分离,没有特别的限制。优选地,(vi)包括,更优选由以下组成:
(vi.1)优选对由(iv)或(v),优选由(v)获得的悬浮液进行固液分离,从而获得母液和包含沸石材料的固体材料,所述沸石材料具有cha骨架类型和包含si、al、o和h的骨架结构;其中所述固-液分离优选包括过滤和离心中的一种或多种;
(vi.2)优选洗涤由(vi.1)获得的固体材料,从而获得包含具有cha骨架类型和包含si、al、o和h的骨架结构的沸石材料的固体材料;
(vi.3)干燥由(iv)、(v)、(vi.1)或(v.2)获得,优选由(v.2)获得的固体材料,从而获得包含具有cha骨架类型和包含si、al、o和h的骨架结构的沸石材料的固体材料。
关于根据(vi.1)的固液分离,优选进行膜过滤或借助压滤机的过滤或借助离心过滤器的过滤。优选地,其中(vi)包括(vi.2),优选用水,优选用去离子水洗涤由(vi.1)获得的固体材料,优选直至由洗涤获得的洗涤水具有使用ph敏感玻璃电极测得为7-8的ph。根据(vi.3),干燥固体材料优选包括制备悬浮液,优选含水悬浮液,所述悬浮液包含由(iv)、(v)、(vi.1)或(vi.2),优选由(v)、(vi.1)或(vi.2),更优选由(vi.2)获得的固体材料,和使悬浮液快速干燥,优选包括喷雾干燥、喷雾造粒干燥和微波干燥中的一种或多种。根据(vi.3),优选在气体气氛中干燥固体材料,所述气体气氛优选具有50-150℃,更优选60-120℃,更优选70-90℃的温度,其中所述气体气氛优选包括氧气和氮气中的一种或多种,其中更优选所述气体气氛包括,更优选是氧气、空气和贫空气中的一种或多种。
优选地,本发明的方法进一步包括:
(vii)煅烧由(vi)获得的固体材料,从而获得具有cha骨架类型和包含si、al、o和h的骨架结构的沸石材料。
优选地,根据(vii),煅烧固体材料,优选在具有500-675℃,更优选550-650℃,更优选575-625℃的温度的气体气氛中煅烧,其中所述气体气氛优选包括氧气和氮气中的一种或多种,其中更优选气体气氛包括,更优选是氧气、空气和贫空气中的一种或多种。优选地,根据(vii),将固体材料加热至100-200℃的温度,更优选110-190℃的温度,更优选125-175℃的温度,在该温度范围下保持0.5-6小时,优选0.75-4.5小时,更优选1-3小时;加热至500-675℃,更优选550-650℃,更优选575-625℃的温度,并且在该温度范围下保持1-12小时,优选2.5-9小时,更优选3-6小时。
优选地,本发明的方法进一步包括:
(viii)将由(vii)获得的具有cha骨架类型和包含si、al、o和h的骨架结构的沸石材料冷却,优选冷却至15-40℃,优选20-30℃的温度。
此外,本发明的方法可包括:
(ix)对如上获得的具有cha骨架类型和包含si、al、o和h的骨架结构的沸石材料实施离子交换方法,从而获得包含具有cha骨架类型且包含m的沸石材料的混合物。
根据(ix),优选使沸石材料中所含的一种或多种离子非骨架元素发生离子交换,更优选与一种或多种阳离子m进行离子交换,其中所述一种或多种阳离子m为sr、zr、cr、mg、mo、fe、co、ni、cu、zn、ru、rh、pd、ag、os、ir和pt中的一种或多种,优选为sr、cr、mo、fe、co、ni、cu、zn和ag中的一种或多种,更优选为cr、mg、mo、fe、ni、cu、zn和ag中的一种或多种,更优选为mg、mo、fe、ni、cu、zn和ag中的一种或多种,更优选为cu和fe中的一种或多种,更优选cu,并且其中所述一种或多种离子非骨架元素优选包括h和碱金属,所述碱金属优选为li、na、k和cs中的一种或多种,更优选为li、na和k中的一种或多种,更优选为na和k中的一种或多种,更优选为na。此外,(ix)优选包括使具有cha骨架类型和包含si、al、o和h的骨架结构的沸石材料与包含m阳离子的溶液接触,从而获得包含含m沸石材料的混合物。根据(ix)使所述溶液与沸石材料接触可重复至少1次,例如1次、2次或3次。优选地,根据(ix)的使所述溶液与沸石材料接触包括用所述溶液浸渍沸石材料和将所述溶液喷涂到沸石材料上中的一种或多种,优选用所述溶液浸渍沸石材料。
此外,本发明的方法可包括:
(x)将具有cha骨架类型且包含m的沸石材料从由(ix)获得的混合物中分离,其中(x)优选包括:
(x.1)使由(ix)获得的混合物经历固液分离方法,优选包括过滤方法、离心方法或喷雾方法,从而获得具有cha骨架类型且包含m的沸石材料;
(iii.2)优选洗涤由(iii.1)获得的沸石材料;
(iii.3)干燥由(iii.1)或(iii.2),优选由(iii.2)获得的沸石材料。
此外,本发明的方法可包括:
(xi)煅烧由(x)获得的沸石材料,从而获得具有cha骨架类型且包含m的沸石材料。
此外,本发明的方法可包括制备包含沸石材料的模制品,其中所述制备模制品优选包括挤出、压片和喷涂,其中更优选地,模制品具有矩形、三角形、六边形、正方形、椭圆形或圆形横截面,和/或优选为星形、片状、球形、圆柱形、线状或空心圆柱形。
此外,本发明涉及一种可通过或通过上述方法获得的具有cha骨架类型和包含si、al、o和h的骨架结构的沸石材料。
此外,本发明涉及一种具有cha骨架类型和包含si、al、o和h的骨架结构的沸石材料,优选可通过上述方法获得或通过上述方法获得的沸石材料,其中在沸石材料的骨架结构中,以al2o3:sio2摩尔比计算的铝相对于硅的摩尔比为0.001:1至0.5:1,优选为0.01:1至0.1:1,优选为0.02:1至0.05:1,更优选为0.03:1至0.04:1。
优选地,构成沸石材料的晶体具有如参考实施例2.4所述通过sem测得为50-1,500nm,优选为75-1,000nm,更优选为90-150nm的晶体尺寸,其中优选至少50%,更优选至少75%,更优选至少90%的晶体具有处于该范围内的尺寸。优选地,所述沸石材料具有如参考实施例2.2所述测得为至少500m2/g的bet比表面积。优选地,所述沸石材料具有如参考实施例2.6所述测得的27al固体nmr谱,其显示出62.0-54.0ppm,优选为60.0-58.0ppm,更优选为59.9-58.6ppm的共振和峰最大值,并且半高宽为至多7.0ppm,优选为至多5.0ppm,更优选为至多4.0ppm。优选地,沸石材料具有如参考实施例2.7所述测得的29si固体nmr谱,其显示出:
-处于-108.1至-114.5ppm,优选-110.3至-112.2ppm,更优选-110.9至-111.7ppm的第一范围内的共振和峰最大值;
-处于-102.6至-108.1ppm,优选-103.9至-106.3ppm,更优选-104.6至-105.4ppm的第二范围内的共振和峰最大值;
-处于-97.7至-102.6ppm,优选-99.7至-101.9ppm,更优选-100.7至-101.5ppm的第三范围内共振峰,其具有或不具有峰最大值;
其中第二范围的积分与第一范围的积分之比优选为0.25:1至0.45:1,更优选为0.31:1至0.39:1,更优选为0.34:1至0.36:1。
根据本发明,所述沸石材料可优选包含cu和fe中的一种或多种,更优选包含cu。
通常,如上所述的沸石材料可用于每一种合适的目的,作为吸附剂、吸收剂、分子筛、催化活性材料、催化剂或催化剂组分,优选作为催化活性材料、催化剂或催化剂组分。优选的用途包括选择性催化还原废气流,优选来自柴油发动机的废气流中的氮氧化物;将c1化合物转化为一种或多种烯烃,优选将甲醇转化为一种或多种烯烃;或将包含一氧化碳和氢气的合成气转化为一种或多种烯烃。因此,本发明还涉及一种催化剂,优选用于选择性催化还原废气流,优选来自柴油发动机的废气流中的氮氧化物的催化剂,或者用于将c1化合物催化转化为一种或多种烯烃,优选将甲醇转化为一种或多种烯烃或将包含一氧化碳和氢气的合成气转化为一种或多种烯烃的催化剂,所述催化剂包含如上所述的沸石材料。
通过由所示的引用和回引产生的以下实施方案集合和实施方案组合来进一步阐述本发明。特别地,注意到在提及实施方案范围的每种情况下,例如在术语如“根据实施方案1-4中任一项的方法”的上下文中,意味着对本领域技术人员明确公开该范围内的每一个实施方案,即该术语的措辞应被本领域技术人员理解为与“根据实施方案1、2、3和4中任一项的方法”同义。
1.一种制备具有cha骨架类型和包含si、al、o和h的骨架结构的沸石材料的间歇方法,包括:
(i)提供包含具有cha骨架类型和包含si、al、o和h的骨架结构的沸石材料的晶种材料;
(ii)制备包含si源、al源、(i)中提供的晶种材料、包含环烷基铵化合物的cha骨架结构导向剂和水的混合物,
其中所述环烷基铵化合物为包含阳离子r1r2r3r4n+的化合物,其中r1、r2、r3彼此独立地为具有1-6个碳原子的烷基,r4为5-8元环烷基,
其中在混合物中,水相对于以sio2计算的si源和晶种材料中所含的si的摩尔比为5:1至15:1,
其中所述混合物中,以na2o计算的钠相对于以sio2计算的si源和晶种材料中所含的si的摩尔比为0:1至0.1:1;
(iii)将(ii)中制备的混合物在其液态下加热至50-90℃的混合物温度,并将液态混合物在该温度下保持5-100小时;
(iv)在结晶容器中将(iii)的加热混合物加热至190-230℃的混合物温度,并在结晶容器中在自生压力下将混合物保持在该温度下0.5-10小时,从而获得悬浮在其母液中的包含具有cha骨架类型和含有si、al、o和h的骨架结构的沸石材料的固体材料。
2.根据实施方案1的方法,其中根据(i)的提供晶种材料包括通过包括以下步骤的方法制备晶种材料:
(i.1)提供具有cha骨架类型和包含si、al、o和h的骨架结构的沸石材料;
(i.2)制备包含(i.1)中提供的沸石材料和液体的悬浮液;
(i.3)研磨(i.2)中制备的悬浮液。
3.根据实施方案1的方法,其中根据(i)提供的晶种材料可通过或通过包括以下步骤的方法获得:
(i.1)提供具有cha骨架类型和包含si、al、o和h的骨架结构的沸石材料;
(i.2)制备包含(i.1)中提供的沸石材料和液体的悬浮液;
(i.3)研磨(i.2)中制备的悬浮液。
4.根据实施方案2或3的方法,其中根据(i.1)的提供具有cha骨架类型和包含si、al、o和h的骨架结构的沸石材料包括制备具有包含sio2和al2o3的cha型骨架结构的沸石材料,其中所述方法包括以下步骤:
(a)提供包含一种或多种sio2源、一种或多种al2o3源、作为结构导向剂的一种或多种含四烷基铵阳离子r1r2r3r4n+的化合物和一种或多种含四烷基铵阳离子r5r6r7r8n+的化合物的混合物;
(b)使所得混合物结晶,从而获得具有cha型骨架结构的沸石材料;其中r1、r2、r3、r4、r5、r6和r7彼此独立地表示烷基,并且其中r8表示环烷基;
其中r1、r2、r3、r4、r5、r6和r7彼此独立地优选表示任选取代和/或任选支化的c1-c6烷基,其中r8优选表示任选杂环和/或任选取代的5-8元环烷基,其中结晶优选在溶剂热条件下进行,其中步骤(1)中提供的混合物不含任何显著量的含三甲基苄基铵的化合物,其中步骤(a)中提供的混合物进一步包含晶种。
5.根据实施方案2-4中任一项的方法,其中根据(i.1)提供的沸石材料优选为可通过或通过实施方案4的方法获得的沸石材料,其中所述沸石材料优选表现出以下参数中的一个或多个:
-3,720-3,740cm-1的第一吸收带(b1)和1,850-1,890cm-1的第二吸收带(b2);
其中第一吸收带的最大吸光度与第二吸收带的最大吸光度之比b1:b2为0.5-1.55;
-所述沸石材料的粒度dv10为400-2,500nm;
-所述沸石材料的粒度dv50为600-3,500nm;
-所述沸石材料的粒度dv90为1,200-4,500nm;
-不含任何显著量的元素p和/或as;
-所述沸石材料的29simasnmr包含-102.0至-106.0ppm的第一峰(p'1)和-108.0至-112.5ppm的第二峰(p'2);其中所述沸石材料的29simasnmr中的第一峰和第二峰的积分提供了0.05-0.90的积分值之比p'1:p'2;
-沸石骨架结构的sio2:al2o3摩尔比为4:1至200:1。
6.根据实施方案2-5中任一项的方法,其中根据(i.2)的制备悬浮液包括将(i.1)中提供的沸石材料与液体混合,其中所述液体优选包含水,更优选包含95-100重量%,更优选99-100重量%的水(如果所述液体由水组成),且其中沸石材料相对于液体的重量比优选为1:10至1:50,优选为1:20至1:40,更优选为1:25至1:35。
7.根据实施方案2-6中任一项的方法,其中根据(i.3)研磨悬浮液包括珠磨悬浮液,优选持续10-240分钟,更优选20-200分钟,更优选30-150分钟的时间,其中珠粒具有优选为100-500微米,更优选为200-400微米,更优选为250-350微米的直径,并且其中珠磨以1,000-5,000rpm,优选2,000-4,000rpm,更优选2,500-3,500rpm操作。
8.根据实施方案2-7中任一项的方法,其中制备晶种材料的方法由(i.1)、(i.2)和(i.3)组成。
9.根据实施方案2-8中任一项的方法,其中根据(i)的提供晶种材料进一步包括,在(i.3)之后,
(i.4)将由(i.3)获得的研磨悬浮液中所含的沸石材料与液体分离。
10.根据实施方案9的方法,其中根据(i.4)的将沸石材料与液体分离包括对由(i.3)获得的经研磨悬浮液进行固液分离,优选包括过滤和离心中的一种或多种,从而获得经分离的沸石材料,和任选地在温度为20-100℃的气体气氛中干燥经分离的沸石材料,其中所述气体气氛优选包括氧气和氮气中的一种或多种。
11.根据实施方案2-9中任一项的方法,其中制备晶种材料的方法由(i.1)、(i.2)、(i.3)和(i.4)组成。
12.根据实施方案1-11中任一项的方法,其中在(ii)中制备的混合物中,晶种材料相对于以sio2计算的si源中所含的si的重量比为0.025:1至0.15:1,优选为0.030:1至0.13:1,更优选为0.035:1至0.11:1。
13.根据实施方案1-12中任一项的方法,其中在(ii)中制备的混合物中,晶种材料相对于以sio2计算的si源中所含的si的重量比为0.025:1至0.060:1,优选为0.030:1至0.055:1,更优选为0.035:1至0.045:1。
14.根据实施方案1-12中任一项的方法,其中在(ii)中制备的混合物中,晶种材料相对于以sio2计算的si源中所含的si的重量比为0.085:1至0.15:1,优选为0.090:1至0.13:1,更优选为0.095:1至0.11:1。
15.根据实施方案1-14中任一项的方法,其中r1、r2、r3彼此独立地为具有1-5个碳原子,优选1-4个碳原子,更优选1-3个碳原子,更优选1或2个碳原子,更优选1个碳原子的烷基,且其中r4为5-7元环烷基,优选5或6元环烷基,更优选6元环烷基。
16.根据实施例1-15中任一项的方法,其中r1、r2、r3为甲基,r4为环己基。
17.根据实施方案1-16中任一项的方法,其中根据(ii)的cha骨架结构导向剂中所含的环烷基铵化合物包括,优选是铵盐,优选卤化物、硫酸盐、硝酸盐、乙酸盐和氢氧化物中的一种或多种,更优选氯化物、溴化物、硫酸盐、硝酸盐、乙酸盐和氢氧化物中的一种或多种,更优选氯化物、溴化物和氢氧化物中的一种或多种,更优选氢氧化物。
18.根据实施方案1-17中任一项的方法,其中根据(ii)的cha骨架结构导向剂中所含的环烷基铵化合物包括,优选是n,n,n-三甲基环己基氢氧化铵。
19.根据实施方案1-18中任一项的方法,其中99-100mol%,优选99.5-100mol%,更优选99.9-100mol%的cha骨架结构导向剂由n,n,n-三甲基环己基氢氧化铵组成,其中更优选cha骨架结构导向剂不含n,n,n-三甲基-1-金刚烷基氢氧化铵,优选不含含n,n,n-三甲基-1-金刚烷基铵的化合物,更优选不含含金刚烷基铵的化合物。
20.根据实施方案1-19中任一项的方法,其中根据(ii)的cha骨架结构导向剂进一步包括四烷基铵化合物,所述四烷基铵化合物包含阳离子r5r6r7r8n+,其中r5、r6、r7、r8彼此独立地为任选取代的具有1-4个碳原子,优选1-3个碳原子,更优选1或2个碳原子的烷基,其中更优选r5、r6、r7、r8为甲基,其中取代基优选为氯和羟基中的一种或多种,更优选为羟基。
21.根据实施方案20的方法,其中根据(ii)的cha骨架结构导向剂中所含的四烷基铵化合物包括,优选是铵盐,优选卤化物、硫酸盐、硝酸盐、乙酸盐和氢氧化物中的一种或多种,更优选氯化物、溴化物、硫酸盐、硝酸盐、乙酸盐和氢氧化物中的一种或多种,更优选氯化物、溴化物和氢氧化物中的一种或多种,更优选氢氧化物。
22.根据实施方案20或21的方法,其中根据(ii)的cha骨架结构导向剂中所含的四烷基铵化合物优选包括,优选是四甲基氢氧化铵。
23.根据实施方案20-22中任一项的方法,其中在根据(ii)的cha骨架结构导向剂中,环烷基铵化合物相对于四烷基铵化合物的摩尔比为1:1至5.5:1,优选为1.1:1至4:1,更优选为1.3:1至3:1,更优选为1.5:1至2.0:1。
24.根据实施方案20-23中任一项的方法,其中99-100mol%,优选99.5-100mol%,更优选99.9-100mol%的cha骨架结构导向剂由环烷基铵化合物和四烷基铵化合物组成,其中更优选cha骨架结构导向剂不含n,n,n-三甲基-1-金刚烷基氢氧化铵,优选不含含n,n,n-三甲基-1-金刚烷基铵的化合物,更优选不含含金刚烷基铵的化合物。
25.根据实施方案1-24中任一项的方法,其中在(ii)中制备的混合物中,cha骨架结构导向剂相对于以sio2计算的si源和晶种材料中所含的si的摩尔比为0.20:1至0.30:1,优选为0.22:1至0.29:1,更优选为0.25:1至0.28:1。
26.根据实施方案1-25中任一项的方法,其中在(ii)中制备的混合物中,水相对于以sio2计算的si源和晶种材料中所含的si的摩尔比为7:1至15:1,优选为9:1至12:1。
27.根据实施方案1-26中任一项的方法,其中si源包括,优选是二氧化硅和硅酸盐中的一种或多种,优选热解法二氧化硅、硅溶胶、无定形二氧化硅、硅胶、硅酸、硅酸酯、胶态二氧化硅、四烷氧基硅烷、二硅酸盐和倍半硅酸盐中的一种或多种,更优选热解法二氧化硅、二氧化硅水溶胶、硅胶、硅酸、硅酸酯、胶态二氧化硅和四烷氧基硅烷中的一种或多种,更优选热解法二氧化硅、二氧化硅水溶胶、硅胶和胶态二氧化硅中的一种或多种,更优选热解法二氧化硅、硅胶和胶态二氧化硅中的一种或多种。
28.根据实施方案1-27中任一项的方法,其中si源包括,更优选是胶态二氧化硅。
29.根据实施方案1-28中任一项的方法,其中al源包括,优选是氧化铝、铝酸盐和铝盐中的一种或多种,优选氧化铝和铝盐中的一种或多种,更优选氧化铝、三(c1-c5)醇盐、alo(oh)、al(oh)3、卤化铝(其中卤化铝优选氟化铝、氯化铝和溴化铝中的一种或多种)、硫酸铝、磷酸铝和氟硅酸铝中的一种或多种,更优选alo(oh)和al(oh)3中的一种或多种。
30.根据实施方案1-29中任一项的方法,其中al源包括,更优选是al(oh)3,优选结晶al(oh)3,更优选三水铝石。
31.根据实施方案1-30中任一项的方法,其中在(ii)中制备的混合物中,以al2o3计算的al源相对于以sio2计算的si源和晶种材料中所含的si的摩尔比为0.001:1至0.5:1,优选为0.01:1至0.1:1,优选为0.02:1至0.05:1,更优选为0.03:1至0.04:1。
32.根据实施方案1-31中任一项的方法,其中95-100重量%,优选98-100重量%,更优选99-100重量%的(ii)中制备的混合物由si源、al源、(i)中提供的晶种材料、包含环烷基铵化合物的cha骨架结构导向剂和水组成。
33.根据实施方案1-32中任一项的方法,其中99.5-100重量%,优选99.9-100重量%,更优选99.9-100重量%的(ii)中制备的混合物由si源、al源、(i)中提供的晶种材料、包含环烷基铵化合物的cha骨架结构导向剂和水组成。
34.根据实施方案1-33中任一项的方法,其中在(ii)中制备的混合物中,以元素p计算的磷相对于以sio2计算的si源中和晶种材料中所含的si的摩尔比为0:1至0.001:1。
35.根据实施方案1-34中任一项的方法,其中根据(ii)的制备混合物包括:
(ii.1)制备包含si源、al源、包含环烷基铵化合物的cha骨架结构导向剂和水的混合物;
(ii.2)将晶种材料加入到(ii.1)中制备的混合物中,从而获得待实施(iii)的混合物。
36.根据实施方案35的方法,其中(ii.1)包括:
(ii.1.1)制备包含al源和cha骨架结构导向剂的混合物,所述cha骨架结构导向剂包含环烷基铵化合物;
(ii.1.2)将(ii.1.1)中制备的混合物在10-50℃的混合物温度下搅动5-60分钟;
(ii.1.3)将si源加入到由(ii.1.2)获得的混合物中;
(ii.1.4)将(ii.1.3)中制备的混合物在10-50℃的混合物温度下搅动1-30分钟,从而获得待实施(iii)的混合物。
37.根据实施方案36的方法,其中根据(ii.1.2),将混合物在15-40℃,优选20-30℃的混合物温度下搅动。
38.根据实施方案36或37的方法,其中根据(ii.1.2),将混合物搅动10-50分钟,优选20-40分钟。
39.根据实施方案36-38中任一项的方法,其中根据(ii.1.2),将混合物在0.7-2巴(绝对),优选0.8-1.5巴(绝对),更优选0.9-1.1巴(绝对)的压力下搅动。
40.根据实施方案36-39中任一项的方法,其中根据(ii.1.2)的搅动包括机械搅动混合物,优选搅拌混合物。
41.根据实施方案40的方法,其中根据(ii.1.2),将混合物在100-1,000rpm,优选200-750rpm,更优选400-600rpm下搅动。
42.根据实施方案36-41中任一项的方法,其中根据(ii.1.4),将混合物在15-40℃,优选20-30℃的混合物温度下搅动。
43.根据实施方案36-42中任一项的方法,其中根据(ii.1.4),将混合物搅动2-20分钟,优选5-15分钟。
44.根据实施方案36-43中任一项的方法,其中根据(ii.1.4),将混合物在0.7-2巴(绝对),优选0.8-1.5巴(绝对),更优选0.9-1.1巴(绝对)的压力下搅动。
45.根据实施方案36-44中任一项的方法,其中根据(ii.1.4)的搅动包括机械搅动所述混合物,优选搅拌所述混合物。
46.根据实施方案45的方法,其中根据(ii.1.4),将混合物在100-1,000rpm,优选200-750rpm,更优选400-600rpm下搅拌。
47.根据实施方案36-46中任一项的方法,其中(ii.1)由(ii.1.1)、(ii.1.2)、(ii.1.3)和(ii.1.4)组成。
48.根据实施方案35-47中任一项的方法,其中(ii)由(ii.1)和(ii.2)组成。
49.根据实施方案1-48中任一项的方法,其中根据(iii),将(ii)制备的混合物在其液态下加热至55-80℃,优选60-70℃的混合物温度。
50.根据实施方案1-49中任一项的方法,其中根据(iii),将液态混合物在该温度范围下保持5-80小时,优选20-50小时。
51.根据实施方案1-50中任一项的方法,其中根据(iii)的加热混合物,优选根据(iii)的加热混合物并将混合物保持在该温度下在0.7-2巴(绝对),优选0.8-1.5巴(绝对),更优选0.9-1.1巴(绝对)的压力下进行。
52.根据实施方案1-51中任一项的方法,其中根据(iii),将混合物以0.2-5k/分钟,优选0.5-4k/分钟,更优选1-3k/分钟的升温速率下加热至该温度。
53.根据实施方案1-52中任一项的方法,其中在根据(iii)的将混合物保持该温度下期间,优选在根据(iii)的加热混合物并将混合物保持在该温度下期间,搅动混合物。
54.根据实施方案53的方法,其中根据(iii)的搅动包括机械搅动混合物,优选搅拌混合物。
55.根据实施方案54的方法,其中根据(iii),将混合物在100-1,000rpm,优选200-750rpm,更优选400-600rpm下搅拌。
56.根据实施方案1-55中任一项的方法,其中(iii)由加热混合物和将混合物保持在该温度下组成。
57.根据实施方案1-56中任一项的方法,其中根据(iv),将(iii)的混合物加热至195-225℃,优选200-220℃的混合物温度。
58.根据实施方案1-57中任一项的方法,其中根据(iv),将混合物在该温度下保持0.75-7.5小时,优选1-5小时。
59.根据实施方案1-58中任一项的方法,其中根据(iv),将混合物以0.1-20k/分钟,优选0.5-15k/分钟,优选1-10k/分钟的升温速率加热至所述温度。
60.根据实施方案1-59中任一项的方法,其中在根据(iv)的加热混合物或将混合物保持该温度下期间,优选在根据(iv)的加热混合物并将混合物保持在该温度下期间,搅动混合物,优选机械搅动,其中更优选搅动结晶容器。
61.根据实施方案1-60中任一项的方法,其中根据(iv)的结晶容器为高压釜。
62.根据实施方案1-60中任一项的方法,其中根据(iv)的结晶容器是包括反应管和一个或两个用于密封反应管的密封盖的管式反应器。
63.根据实施方案62的方法,其中在根据(iv)的加热混合物并将混合物保持在该温度下期间,通过加热介质外部加热管式反应器。
64.根据实施方案63的方法,其中加热介质为气态加热介质、液态加热介质或固态加热介质,优选为气态加热介质或液态加热介质。
65.根据实施方案64的方法,其中加热介质为气态加热介质,优选包含在静态或连续操作的烘箱中,其中,在根据(iv)的加热和保持在该温度下期间,使气态加热介质静态或连续地与反应管接触。
66.根据实施方案64的方法,其中加热介质为液态加热介质,优选包括油,所述加热介质更优选包含在静态或连续操作的浴中,其中在根据(iv)的加热和保持在该温度期间,使液态加热介质静态或连续地与反应管接触。
67.根据实施方案62-66中任一项的方法,其中反应管由热扩散系数为3×10-6至30×10-6m2/s,优选为5×10-6至25×10-6m2/s的材料制成。
68.根据实施方案67的方法,其中所述材料是不锈钢。
69.根据实施方案62-68任一项的方法,其中反应管的体积v/cm3与反应管的外表面积a/cm2之比v/a为0.1:1至100:1,优选为0.2:1至60:1,更优选为0.5:1至10:1。
70.根据实施方案62至69中任一项的方法,其中(iv)包括,优选由以下组成:
(iv.1)将由(iii)获得的经加热的混合物供应至反应管;
(iv.2)用所述一个或两个密封盖密封反应管;
(iv.3)加热(iii)的加热混合物并将混合物保持在该温度下,从而获得悬浮在
其母液中的包含具有cha骨架类型和包含si、al、o和h的骨架结构的沸石材料的固体材料;
(iv.4)任选地冷却由(iv.3)获得的悬浮液;
(iv.5)打开一个或两个密封盖,从反应管中移出由(iv.3)或(iv.4)中获得的悬浮液。
71.根据实施方案70的方法,其中在根据(iv.3)的加热期间,加热介质的温度为190-235℃,优选为195-230℃,更优选为200-225℃。
72.根据实施方案1-71中任一项的方法,进一步包括:
(v)将由(iv)获得的悬浮液冷却,优选冷却至15-40℃,优选20-30℃的温度。
73.根据实施方案1-72中任一项的方法,进一步包括:
(vi)将由(iv)或(v),优选由(v)获得的包含有cha骨架类型和包含si、al、o和h的骨架结构的沸石材料的固体材料与其母液分离,从而获得包含具有cha骨架类型和包含si、al、o和h的骨架结构的沸石材料的固体材料。
74.根据实施方案73的方法,其中(vi)包括以下步骤,优选由以下步骤组成:
(vi.1)优选对由(iv)或(v),优选由(v)获得的悬浮液进行固液分离,从而获得母液和包含沸石材料的固体材料,所述沸石材料具有cha骨架类型和包含si、al、o和h的骨架结构;
(vi.2)优选洗涤由(vi.1)获得的固体材料,从而获得包含具有cha骨架类型和包含si、al、o和h的骨架结构的沸石材料的固体材料;
(vi.3)干燥由(iv)、(v)、(vi.1)或(v.2)获得的固体材料,优选由(v.2)获得的固体材料,从而获得包含具有cha骨架类型和包含si、al、o和h的骨架结构的沸石材料的固体材料。
75.根据实施方案74的方法,其中(vi)包括(vi.1),其中根据(vi.1)的固液分离包括过滤和离心中的一种或多种。
76.根据实施方案74或75的方法,其中(vi)包括(vi.2),其中将由(vi.1)获得的固体材料用水,优选用去离子水洗涤,优选直至由洗涤获得的洗涤水具有使用ph敏感玻璃电极测得为7-8的ph。
77.根据实施方案74-76中任一项的方法,其中根据(vi.3),干燥固体材料包括制备悬浮液,优选含水悬浮液,其包含由(iv)、(v)、(vi.1)或(vi.2),优选由(v)、(vi.1)或(vi.2),更优选由(vi.1)或(vi.2),更优选由(vi.2)获得的固体材料,和对悬浮液进行快速干燥,所述快速干燥优选包括喷雾干燥、喷雾造粒干燥和微波干燥中的一种或多种。
78.根据实施方案74-77中任一项的方法,其中根据(vi.3),将固体材料在气体气氛中干燥,所述气体气氛优选具有50-150℃,更优选60-120℃,更优选70-90℃的温度,其中所述气体气氛优选包括氧气和氮气中的一种或多种,其中更优选气体气氛包括,更优选是氧气、空气和贫空气中的一种或多种。
79.根据实施方案73-78中任一项的方法,进一步包括:
(vii)煅烧由(vi)获得的固体材料,从而获得具有cha骨架类型和包含si、al、o和h的骨架结构的沸石材料。
80.根据实施方案79的方法,其中根据(vii),将固体材料在气体气氛中煅烧,所述气体气氛优选具有500-675℃,更优选550-650℃,更优选575-625℃的温度,其中所述气体气氛优选包括氧气和氮气中的一种或多种,其中更优选所述气体气氛包括,更优选是氧气、空气和贫空气中的一种或多种。
81.根据实施方案80的方法,其中根据(vii),将固体材料加热至100-200℃,优选110-190℃,更优选125-175℃的温度,在该温度范围下保持0.5-6小时,优选0.75-4.5小时,更优选1-3小时;加热至500-675℃,更优选550-650℃,更优选575至625℃的温度,并在该温度范围下保持1-12小时,优选2.5-9小时,更优选3-6小时。
82.根据实施方案79-81中任一项的方法,进一步包括:
(viii)将由(vii)获得的具有cha骨架类型和包含si、al、o和h的骨架结构的沸石材料冷却,优选冷却至15-40℃,优选20-30℃的温度。
83.根据实施方案82的方法,进一步包括:
(ix)对根据实施方案79-82中任一项的具有cha骨架类型和包含si、al、o和h的骨架结构的沸石材料,优选根据实施方案82的沸石材料实施离子交换方法,从而获得包含具有cha骨架类型且包含m的沸石材料的混合物。
84.根据实施方案83的方法,其中根据(ix),对沸石材料中所含的一种或多种离子非骨架元素实施离子交换,优选与一种或多种阳离子m进行离子交换,其中所述一种或多种阳离子m为sr、zr、cr、mg、mo、fe、co、ni、cu、zn、ru、rh、pd、ag、os、ir和pt中的一种或多种阳离子,优选sr、cr、mo、fe、co、ni、cu、zn和ag中的一种或多种阳离子,更优选cr、mg、mo、fe、ni、cu、zn和ag中的一种或多种阳离子,更优选mg、mo、fe、ni、cu、zn和ag中的一种或多种阳离子,更优选cu和fe中的一种或多种阳离子,更优选cu的阳离子,并且其中所述一种或多种离子非骨架元素优选包括h和碱金属,所述碱金属优选为li、na、k和cs中的一种或多种,更优选li、na和k中的一种或多种,更优选na和k中的一种或多种,更优选为na。
85.根据实施方案83或84的方法,其中(ix)包括使具有cha骨架类型和包含si、al、o和h的骨架结构的沸石材料与包含m的阳离子的溶液接触,从而获得包含m的沸石材料的混合物。
86.根据实施方案85的方法,其中将根据(ix)的使溶液与沸石材料接触重复至少一次。
87.根据实施方案85或86的方法,其中根据(ix)的使溶液与沸石材料接触包括用溶液浸渍沸石材料和将溶液喷雾到沸石材料上中的一种或多种,优选用溶液浸渍沸石材料。
88.根据实施方案83-87中任一项的方法,进一步包括:
(x)从由(ix)获得的混合物中分离具有cha骨架类型且包含m的沸石材料。
89.根据实施方案88的方法,其中根据(x)分离沸石材料包括:
(x.1)对由(ix)获得的混合物实施固液分离方法,优选包括过滤方法、离心方法或喷雾方法,从而获得具有cha骨架类型且包含m的沸石材料;
(iii.2)优选洗涤由(iii.1)获得的沸石材料;
(iii.3)干燥由(iii.1)或(iii.2),优选由(iii.2)获得的沸石材料。
90.根据实施方案89的方法,进一步包括:
(xi)煅烧由(x)获得的沸石材料,从而获得具有cha骨架类型且包含m的沸石材料。
91.根据实施方案1-90中任一项的方法,进一步包括制备包含沸石材料的模制品,所述制备模制品优选包括挤出、压片和喷涂,其中更优选地,所述模制品具有矩形、三角形、六边形、正方形、椭圆形或圆形横截面,和/或优选地为星形、片状、球形、圆柱形、线状或中空圆柱形。
92.可通过或通过根据实施方案1-90中任一项的方法获得的具有cha骨架类型和包含si、al、o和h的骨架结构的沸石材料。
93.一种具有cha骨架类型和包含si、al、o和h的骨架结构的沸石材料,优选根据实施方案92的沸石材料,其中在沸石材料的骨架结构中,以al2o3:sio2摩尔比计算的铝相对于硅的摩尔比为0.001:1至0.5:1,优选为0.01:1至0.1:1,更优选为0.02:1至0.05:1,更优选为0.03:1至0.04:1。
94.根据实施方案92或93的沸石材料,其可通过或通过根据实施方案1-82中任一项的方法获得,其中所述方法优选由(i)、(ii)、(iii)、(iv)、(v)、(vi)、(vii)和优选的(viii)组成。
95.根据实施方案92-94中任一项的沸石材料,其中构成沸石材料的晶体具有通过参考实施例2.4中所述的sem测得为50-1,500nm,优选为75-1,000nm,更优选为90-150nm的晶体尺寸,其中优选至少50%,更优选至少75%,更优选至少90%的晶体具有处于该范围内的尺寸。
96.根据实施方案92-95中任一项的沸石材料,其具有如参考实施例2.2所述测得为至少500m2/g的bet比表面积。
97.根据实施方案92-96中任一项的沸石材料,其具有如参考实施例2.6所述测定的27al固体nmr谱,其显示出处于62.0-54.0ppm,优选60.0-58.0ppm,更优选59.9-58.6ppm范围内的共振和峰最大值,并且具有至多7.0ppm,优选至多5.0ppm,更优选至多4.0ppm的半高全宽。
98.根据实施方案92-97中任一项的沸石材料,其具有如参考实施例2.7所述测定的29si固体nmr谱,其显示出:
-处于-108.1至-114.5ppm,优选-110.3至-112.2ppm,更优选-110.9至-111.7ppm的第一范围内的共振和峰最大值;
-处于-102.6至-108.1ppm,优选-103.9至-106.3ppm,更优选-104.6至-105.4ppm的第二范围内的共振和峰最大值;
-处于-97.7至-102.6ppm,优选-99.7至-101.9ppm,更优选-100.7至-101.5ppm的第三范围内的共振峰,其具有或不具有峰最大值;
其中第二范围的积分与第一范围的积分之比优选为0.25:1至0.45:1,更优选为0.31:1至0.39:1,更优选为0.34:1至0.36:1。
99.根据实施方案92或93的沸石材料,其可通过或通过根据实施方案83-90中任一项的方法获得,其中所述方法优选由(i)、(ii)、(iii)、(iv)、(v)、(vi)、(vii),优选(viii)、(ix),优选(x)组成。
100.根据实施方案99的沸石材料,其包含cu和fe中的一种或多种,优选cu。
101.根据实施方案92-100中任一项的沸石材料作为吸附剂、吸收剂、分子筛、催化活性材料、催化剂或催化剂组分,优选作为催化活性材料、催化剂或催化剂组分的用途。
102.根据实施方案101的用途,用于选择性催化还原废气流,优选来自柴油发动机的废气流中的氮氧化物。
103.根据实施方案101的用途,用于将c1化合物转化为一种或多种烯烃,优选用于将甲醇转化为一种或多种烯烃或将包含一氧化碳和氢气的合成气转化为一种或多种烯烃。
104.一种选择性催化还原废气流,优选来自柴油发动机的废气流中的氮氧化物的方法,所述方法包括使所述废气流与包含根据实施方案92-100中任一项,优选根据实施方案99和100的沸石材料的催化剂接触。
105.一种选择性催化还原废气流,优选来自柴油发动机的废气流中的氮氧化物的方法,所述方法包括通过根据实施方案1-90中任一项,优选根据实施方案83-90中任一项的方法制备沸石材料,并使所述废气流与包含所述沸石材料的催化剂接触。
106.一种将c1化合物催化转化为一种或多种烯烃,优选将甲醇转化为一种或多种烯烃或将包含一氧化碳和氢气的合成气转化为一种或多种烯烃的方法,所述方法包括使所述c1化合物与包含根据实施方案92-100中任一项的沸石材料的催化剂接触。
107.一种将c1化合物催化转化为一种或多种烯烃,优选将甲醇转化为一种或多种烯烃或将包含一氧化碳和氢气的合成气转化为一种或多种烯烃的方法,所述方法包括通过根据实施方案1-90中任一项的方法制备沸石材料,并使所述c1化合物与包含所述沸石材料的催化剂接触。
108.一种催化剂,优选用于选择性催化还原废气流,优选来自柴油发动机的废气流中的氮氧化物的催化剂,或用于将c1化合物催化转化为一种或多种烯烃的催化剂,优选用于将甲醇转化为一种或多种烯烃的催化剂,或用于将包含一氧化碳和氢气的合成气转化为一种或多种烯烃的催化剂,所述催化剂包含根据实施方案92-100中任一项的沸石材料。
通过以下参考实施例、实施例和对比实施例进一步阐述本发明。
实施例
参考实施例1:制备具有cha骨架类型的沸石晶种材料
根据wo2015/185625a1第51页的实施例3所述的常规合成程序制备沸石晶种材料。
作为用于该合成的晶种材料,所用的沸石材料根据以下方法制备:将2,040kg水置于搅拌容器中,并在搅拌下向其中加入3,924kg1-金刚烷基三甲基氢氧化铵溶液(20重量%水溶液)。然后加入415.6kg氢氧化钠溶液(20重量%水溶液),随后加入679kg三异丙醇铝(
所得材料具有558m2/g的bet比表面积和通过粉末x射线衍射测得为105%的结晶度。测得产物的钠含量为0.75重量%,以na2o计算。所述沸石材料具有34:1的二氧化硅:氧化铝摩尔比(sio2:al2o3)。
参考实施例2.1:颗粒分布dv的测定
样品的基于体积的粒度分布dv通过将0.1g沸石粉末分散在100gh2o中并通过超声处理10分钟来进行。动态光散射在具有malvernzetasizer软件6.34版的zetasizernanozs上进行,对每个样品进行5轮10秒的测量时间。所给的值是以纳米计的数均粒度。
参考实施例2.2:bet比表面积的测定
氧化铝的bet比表面积根据din66131或din-iso9277使用液氮测定。
参考实施例2.3:xrd图谱和结晶度的测定
粉末x-射线衍射(xrd)图谱使用装备有d/texultra探测器的衍射仪(rigakuultimaiv)进行,其在40kv和40ma下用cukα单色化辐射操作。扫描步长为0.02°,扫描速率为20°/分钟。结晶度使用20-35°2θ内的峰的积分峰面积计算。
参考实施例2.4:sem照片的测定
在碳胶带上的粉末样品上涂覆os之后,在jsm-7500fa(jeol)上观察场致发射扫描电子显微镜(fe-sem)照片。
参考实施例2.5:icp的测定(元素分析)
在将产物溶解在氢氧化钾溶液中之后,在thermoscientificicap-6300电感耦合等离子体原子发射光谱仪(icp-aes)上进行元素分析(si/al比)。典型地,将约10mg样品溶解在1ml50%氢氧化钾溶液中,然后用去离子水稀释至100ml的最终体积。
参考实施例2.6:27almas固态nmr的测定
在9.4特斯拉、10khz魔角旋转下,采用15°单脉冲采集序列以0.5秒重复时间记录27al固态nmr谱2小时。在测量之前,将样品在62%相对湿度下储存至少60小时。共振间接参比d2o中的al(no3)3,1.1mol/kg,作为零参比,频率为0.26056859,按统一的位移标度,根据iupac推荐2008(pureappl.chem.,第80卷,第1期,第59-84页,2008),使用外部二级标准。参考实施例2.7:29simas固态nmr的测定
在7特斯拉、5khz魔角旋转下,采用在采集期间具有异核射频质子去耦的90°单脉冲采集序列和120秒重复时间记录29si固态nmr谱16小时。在测量之前,将样品在62%相对湿度下储存至少60小时。共振间接参比cdcl3中的me4si,体积分数1%,作为零参比,频率0.19867187,按统一的位移标度,根据iupac推荐2008(pureappl.chem.,第80卷,编号1,第59-84页,2008),使用外部二级标准。
参考实施例2.8:ir光谱
ir光谱是由不含载体材料的样品获得的,其中在测量之前,将所述样品在300℃下在高真空下加热3小时。在具有caf2窗口的高真空测量单元中使用nicolet6700光谱仪进行测量。将得到的数据变换为吸光度值,在基线校正后,对光谱进行分析。
对比实施例1:使用异丙醇铝作为al源,在高压釜中不进行陈化的常规方法
首先混合1.991gn,n,n-三甲基环己基氢氧化铵(chtmaoh)水溶液(20重量%)和0.655g四甲基氢氧化铵(tmaoh)水溶液(25重量%)。然后,在室温下以500rpm搅拌30分钟下,缓慢加入0.295g异丙醇铝。在异丙醇铝溶解后,加入3.020g胶态二氧化硅(
在48小时的合成时间后获得的材料的sem示于图5中。在20小时、24小时和48小时后获得的材料的xrd示于图6中。如参考实施例2.3所述测定的结晶度在20小时的结晶时间后仅为17.2%(底部曲线),在24小时的结晶时间后仅为27.3%(中间曲线)。仅在48小时的结晶时间后,才获得约100%的合理结晶度(顶部曲线)。
对比实施例2:使用异丙醇铝作为al源,在高压釜中不进行陈化的常规方法
首先混合1.998gchtmaoh水溶液(20重量%)和0.656gtmaoh水溶液(25重量%)。然后,在搅拌下缓慢加入0.295g异丙醇铝。在500rpm搅拌30分钟下溶解异丙醇铝之后,加入3.008g
在48小时的合成时间后获得的材料的sem示于图7中。在3小时、5小时、16小时和20小时后获得的材料的xrd示于图8中。如参考实施例2.3所述测定的结晶度在3小时的结晶时间后仅为1.7%,在5小时的结晶时间后为11.7%(底部曲线)。仅在16小时和20小时的结晶时间之后,才获得约100%的合理结晶度(中间曲线和顶部曲线)。
对比实施例3:使用氢氧化铝作为al源,在高压釜中不进行陈化的常规方法
首先混合1.997gchtmaoh水溶液(20重量%)和0.656gtmaoh水溶液(25重量%)。然后,在搅拌下缓慢加入0.112g氢氧化铝(三水铝石)。在室温下以500rpm搅拌混合物30分钟后,加入3.002g
在48小时的合成时间后获得的材料的sem示于图9中。在3小时、5小时、16小时和20小时后获得的材料的xrd示于示于图10中。如参考实施例2.3所述测定的结晶度在3小时的结晶时间后仅为7.4%,在5小时的结晶时间后为19.4%(底部曲线)。仅在16小时和20小时的结晶时间之后,才获得约100%的合理结晶度(中间曲线和顶部曲线)。
对比实施例4:使用氢氧化铝作为al源在95℃下陈化的方法
首先混合282.2gchtmaoh(20重量%水溶液)和82.4gtmaoh(25重量%水溶液)。然后,在搅拌下缓慢加入11.2g氢氧化铝(三水铝石)。在室温下以450rpm搅拌混合物过夜后,加入300.4g
发现获得的具有cha骨架类型的沸石材料的结晶度(如参考实施例2.3所述测定)在1.5h的结晶时间后仅为32%,在2h的结晶时间后仅为30%。对比实施例5:使用三异丙基铝作为al源在95℃下陈化的方法
首先混合276.8gchtmaoh(20重量%水溶液)和78.0gtmaoh(25重量%水溶液)。然后,在搅拌下缓慢加入34.8g三异丙基铝。在室温下以450rpm搅拌混合物1.5后,加入358.3g
发现在3小时的结晶时间后,如参考实施例2.3所述测得的具有cha骨架类型的所得沸石材料的结晶度小于10%。
对比实施例6:在95℃下陈化且在陈化之前或之后加入晶种的方法
进行四个独立的间歇实验以评价陈化前后种晶的效果。所有陈化批料的sio2/al2o3/chtmaoh/tmaoh/h2o摩尔比为1/0.036/0.14/0.09/11.5,基于20mmol的sio2。首先混合chtmaoh水溶液(20重量%水溶液)和tmaoh水溶液(25重量%水溶液),随后在500rpm搅拌30分钟下加入al(oh)3,随后加入
如参考实施例2.3所述测定1-4小时的结晶度示于下表1中。
表1.在95℃下陈化1和4天的陈化时间前后的效果
*措辞“之前”和“之后”表示是在95℃下陈化“之前”,将根据参考实施例1制备的晶种加入到预结晶合成混合物中,因此存在于陈化过程中;或者在紧临在95℃下陈化“之后”且在水热处理开始之前,将根据参考实施例1制备的晶种加入到预结晶合成混合物中,因此不存在于陈化过程中,而是存在于结晶过程中。
对比实施例7:1-金刚烷基三甲基氢氧化铵介导的陈化方法与含水n,n,n-三甲基环己基氢氧化铵陈化方法的反应性的比较
以下实验证明了反应性更高的1-金刚烷基三甲基氢氧化铵介导的方法与本发明方法之间的反应性差异。
将结构导向剂1-金刚烷基三甲基氢氧化铵(253.6g,25重量%水溶液)与氢氧化钠水溶液(28.8g,50重量%水溶液)和水(64.8g)混合。加入氢氧化铝(9.3g),将混合物在室温下搅拌30分钟,随后加入
作为比较,首先混合n,n,n-三甲基环己基氢氧化铵水溶液(795.4g,20重量%水溶液)和tmaoh(163.3g,25重量%水溶液),随后在500rpm搅拌30分钟下加入al(oh)3(78g),随后加入
实施例1:研磨晶种材料
使用珠磨装置(lmz015,ashizawafinetechltd.)研磨如参考实施例1所述制备的沸石晶种材料。将10g根据参考实施例1制备的沸石粉末分散在300ml水中,并使用直径为300微米的氧化锆珠,用珠磨装置以3,000rpm研磨120分钟。在容器中,75%的体积填充有氧化锆珠。浆料的最终浓度为7重量%。在研磨处理后,任选地通过离心干燥浆料,并回收残余固体。图1显示了分别制备的沸石晶种材料的xrd,图2显示了所得晶种材料的基于体积的粒度分布(dv),图3和图4显示了该材料的sem照片。
实施例2:在管式反应器中使用作为al源的氢氧化铝和干磨的沸石晶种材料通过陈化制备具有cha骨架类型的沸石材料
首先混合1.994gchtmaoh水溶液(20重量%)和0.669gtmaoh水溶液(25重量%)。然后,在搅拌下缓慢加入0.112g氢氧化铝(三水铝石)。在室温下以500rpm搅拌混合物30分钟后,加入3.008g
在3小时的合成时间后获得的材料的sem示于图11中。在1小时、2小时、3小时和4小时后获得的材料的xrd示于图12中。如参考实施例2.3所述测定的结晶度在2小时的结晶时间后已经为23.4%(从底部数的第二条曲线),在仅3小时的结晶时间后为97%(从顶部数的第二条曲线)。从3小时到4小时结晶,结晶度增加至98%(顶部曲线)。
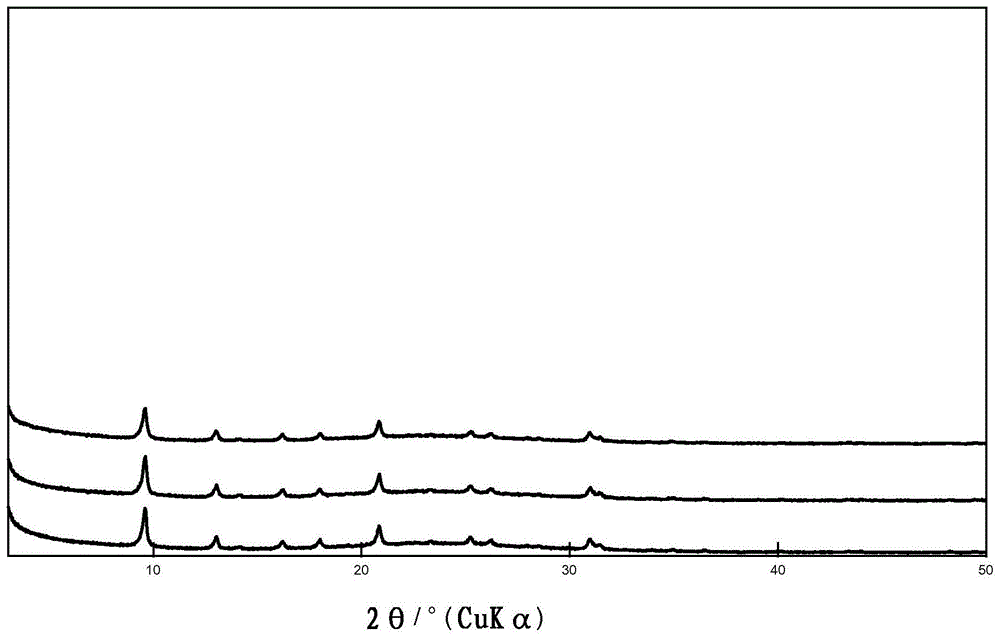
实施例3:在管式反应器中使用作为al源的氢氧化铝和干磨的沸石晶种材料通过陈化制备具有cha骨架类型的沸石材料
首先混合1.996gchtmaoh水溶液(20重量%)和0.663gtmaoh水溶液(25重量%)。然后,在搅拌下缓慢加入0.111g氢氧化铝(三水铝石)。在室温下以500rpm搅拌混合物30分钟后,加入3.007g
在3小时的合成时间后获得的材料的sem示于图13中。在1小时、2小时、3小时和4小时后获得的材料的xrd示于图14中。如参考实施例2.3所述测定的结晶度在2小时的结晶时间后已经为44.6%(从底部数的第二条曲线),在仅3小时的结晶时间后为96%(从顶部数的第二条曲线)。从3小时到4小时的结晶,结晶度增加至100%(顶部曲线)。
实施例4:在高压釜中使用作为al源的氢氧化铝和沸石晶种材料浆料通过陈化制备具有cha骨架类型的沸石材料
首先混合3.989gchtmaoh水溶液(20重量%)和1.313gtmaoh水溶液(25重量%)。然后,在搅拌下缓慢加入0.225g氢氧化铝(三水铝石)。在室温下以500rpm搅拌混合物30分钟后,加入6.006g
元素分析显示si含量为38重量%(以元素计算),al含量为2.8重量%(以元素计算)。在5小时的合成时间后获得的材料的sem照片示于图15中。在5小时后获得的材料的xrd示于图16中,其中6个不同的样品根据该配方制备。如参考实施例2.3所述测定的平均结晶度为95%。
实施例5:在管式反应器中使用作为al源的氢氧化铝和干磨的沸石晶种材料通过陈化制备具有cha骨架类型的沸石材料
首先混合2.510gchtmaoh水溶液(20重量%)和0.836gtmaoh水溶液(25重量%)。然后,在搅拌下缓慢加入0.112g氢氧化铝(三水铝石)。在室温下以500rpm搅拌混合物30分钟后,加入3.004g
在2小时的合成时间后获得的材料的sem示于图17中。在1小时、2小时和3小时后获得的材料的xrd示于图18中。如参考实施例2.3所述测定的结晶度在1小时的结晶时间后已经为66.8%(底部曲线),在仅2小时的结晶时间后为95%(中间曲线)。
实施例6:在管式反应器中使用作为al源的氢氧化铝和干磨的沸石晶种材料通过陈化制备具有cha骨架类型的沸石材料
首先混合3.768gchtmaoh水溶液(20重量%)和1.244gtmaoh水溶液(25重量%)。然后,在搅拌下缓慢加入0.169g氢氧化铝(三水铝石)。在室温下以500rpm搅拌混合物30分钟后,加入4.506g
该材料的二氧化硅:氧化铝摩尔比为26:1。bet比表面积为635m2/g。煅烧材料的sem示于图19中。在仅2小时的结晶时间后,所得材料的结晶度为97%。
该材料的27al固态nmr谱示于图20中。该谱图在59ppm处显示出半高宽为4ppm的主共振,这可归于正交配位的al。在30ppm处观察到强度小于主共振并且没有完全从主共振中分辨出来的共振,这可归于四配位或五配位的al。在-3ppm处观察到强度小于主共振的另一次共振,这可归于八面体配位的al。在-35ppm处观察到主共振的旋转边带。
该材料的29si固态nmr谱示于图21中。该谱图在-111ppm处显示出半高宽为1.8ppm的共振,我们将其归于si(4osi,0oal,0oh)。该谱图在-105ppm处显示出半高全宽为2.8ppm的第二共振,我们将其归于si(3osi,1oal,0oh)。在-97至-103ppm处观察到另一未分辨的共振,其可源自si(2osi,2oal,0oh)或si(3osi,0oal,1oh)。
实施例7:基于根据实施例4制备的沸石材料的催化测试
将根据上述实施例4制备的沸石材料在其干燥和非煅烧状态下用硝酸铜水溶液湿浸渍(初湿浸渍)。然后将该材料干燥并在450℃下煅烧5小时,分别获得具有cha骨架类型的沸石材料,其含有其量为3.5重量%的cu(以cuo计),基于材料的总重量。然后,通过制备其中加入有乙酸锆作为粘合剂材料前体(基于沸石材料为5重量%)的含水浆料而使材料成型。然后将该浆料成型为片剂,在搅拌下干燥并在550℃下煅烧1小时。然后将分别获得的片剂破碎并筛分至250-500微米的粒度。然后将催化剂在650℃下在10%蒸汽/空气中陈化50小时,并在800℃下在10%蒸汽/空气中陈化16小时。通过使催化材料经受气时空速为80,000h-1、气流温度为200℃、400℃、575℃(用于脱生(degreening)的首轮运行),和175℃、200℃、225℃、250℃、300℃、450℃、550℃、575℃的气流(500ppmno、500ppmnh3、5%h2o、10%o2、余量n2)来施加标准scr条件。将催化材料的量调节为每个反应器120mg;用刚玉将材料稀释至约1ml体积。该空速模拟了1ml的涂布催化剂。模拟scr测试的结果示于图22和23中。
附图简介
图1显示了根据实施例1制备的沸石晶种材料的xrd。
图2显示了根据实施例1制备的晶种材料的粒度分布。
图3显示了根据参考实施例2.4获得的根据实施例1制备的晶种材料的sem(放大50,000倍)。
图4显示了根据参考实施例2.4获得的根据实施例1制备的晶种材料的sem(放大75,000倍)。
图5显示了根据参考实施例2.4获得的根据对比实施例1制备的沸石材料的sem(放大30,000倍)。
图6显示了根据对比实施例1制备的沸石材料的xrd图,由下至上:结晶时间=20小时、24小时、48小时。
图7显示了根据参考实施例2.4获得的根据对比实施例2制备的沸石材料的sem(放大25,000倍)。
图8显示了根据对比实施例2制备的沸石材料的xrd图。由下至上:结晶时间=3小时、5小时、16小时、20小时。
图9显示了根据参考实施例2.4获得的根据对比实施例3制备的沸石材料的sem(放大25,000倍)。
图10显示了根据对比实施例3制备的沸石材料的xrd图。由下至上:结晶时间=3小时、5小时、16小时、20小时。
图11显示了根据参考实施例2.4获得的根据实施例2制备的沸石材料的sem(放大75,000倍)。
图12显示了根据实施例2制备的沸石材料的xrd图。由下至上:结晶时间=1小时、2小时、3小时、4小时。
图13显示了根据参考实施例2.4获得的根据实施例3制备的沸石材料的sem(放大75,000倍)。
图14显示了根据实施例3制备的沸石材料的xrd图。由下至上:结晶时间=1小时、2小时、3小时、4小时。
图15显示了根据参考实施例2.4获得的根据实施例4制备的沸石材料的sem(左上放大75,000倍,右上放大50,000倍,左下放大25,000倍,右下放大10,000倍)。
图16显示了根据实施例4制备的6个沸石材料样品在结晶时间为5小时时的xrd图。
图17显示了根据参考实施例2.4获得的根据实施例5制备的沸石材料在2小时的结晶时间后的sem照片(放大75,000倍)。
图18显示了根据实施例5制备的3个沸石材料样品在1小时、2小时和3小时的结晶时间(由下至上)时的xrd图。
图19显示了根据参考实施例2.4获得的根据实施例6制备的沸石材料的sem(放大30,000倍)。
图20显示了根据实施例6制备的材料的27al固态nmr谱。
图21显示了根据实施例6制备的材料的29si固态nmr谱。
图22显示了根据实施例7的催化测试的结果,其中y轴示出了x-nox/%,其中具有向下的三角形
图23显示了根据实施例7的催化测试的结果,其中y轴示出了n2o/ppm,其中具有向上的三角形的曲线(δ)示出了陈化16小时、800℃的催化材料的行为;具有正方形的曲线示出了陈化50小时、650℃的催化材料的行为;具有圆形的曲线示出了新鲜材料的行为。
引用的现有技术
-us4,544,538
-wo2015/185625a
-us20170113941a
- 还没有人留言评论。精彩留言会获得点赞!