金属化合物粒子群、蓄电装置用电极、蓄电装置、及金属化合物粒子群的制造方法与流程
本发明涉及一种金属化合物粒子群、蓄电装置用电极、蓄电装置、及金属化合物粒子群的制造方法。
背景技术:
近年来,随着数码相机、智能手机、便携式个人计算机(personalcomputer,pc)的迅速普及,二次电池的开发变得活跃。二次电池也期待应用于受到燃料价格高涨或对环境负荷的意识提高的影响的汽车动力用途或智能电网(smartgrid)的蓄电用途。
二次电池的电极具有含有可逆地吸藏及放出锂离子的活性物质的正极和负极、及溶解有锂盐的电解液。在此锂离子二次电池中,锂离子通过充电放电从正极放出并吸藏到负极,锂离子通过放电从负极放出并吸藏到正极。锂离子二次电池具有能够在高电压下工作且能量密度大的优点。
另外,为了有效利用锂离子二次电池与电双层电容器两者的优点,提出了所谓混合电容器(hybridcapacitor)。关于混合电容器,将具有电双层电容的分极性电极作为正极,将能够进行锂的吸藏及放出的金属化合物粒子作为负极,并且兼具锂离子二次电池与电双层电容器的优点。即,所述混合电容器具有高能量密度并具有高的输入输出特性。
[现有技术文献]
[专利文献]
[专利文献1]日本专利特开2015-189608号公报
技术实现要素:
[发明所要解决的问题]
在将用作所述蓄电装置的电极材料的金属化合物粒子作为负极材料使用的情况下,由于进行充电放电,有时会在负极上产生堆积物。此堆积物与被称为固体电解质界面(solidelectrolyteinterphase,sei)的皮膜不同,在堆积物产生的时刻导致了内部电阻的增加。
因此,本发明目的在于提供一种可抑制蓄电装置的内部电阻增加的金属化合物粒子群、包含金属化合物粒子群的蓄电装置用电极、及金属化合物粒子群的制造方法。
[解决问题的技术手段]
为了实现所述课题,本实施方式的金属化合物粒子群具有以下结构。
(1)一种金属化合物粒子群,具有金属化合物粒子相连而成的三维网状结构,且在所述金属化合物粒子群的一部分上形成有包含硅氧化物的涂布层。
(2)所述包含硅氧化物的涂布层可形成在所述金属化合物粒子的表面的至少一部分上。
(3)si元素的量相对于金属化合物粒子群整体可为0.4以上3.0wt%以下。
(4)在所述三维网状结构中可存在空隙,在划定所述空隙的所述金属化合物粒子上可形成有所述包含硅氧化物的涂布层。
(5)所述涂布层中含有的硅氧化物可为非晶状的硅氧化物。
(6)可具有相对于10nm以下的细孔径而言为超过0.008dvp/drp的细孔容积变化率,所述细孔容积变化率是根据利用氮气吸附测定法对所述金属化合物粒子群进行测定而得的细孔分布所换算的细孔容积变化率。
(7)相对于未形成所述包含硅氧化物的涂布层的金属化合物粒子群,积分强度的减少率可为25%以上。
(8)所述金属化合物粒子可为钛酸锂。
也可制成包含所述金属化合物粒子群的蓄电装置用电极。另外,也可制成蓄电装置,所述蓄电装置包括:具有作为蓄电装置用电极的负极及正极的元件;以及含浸在所述元件中的电解液,所述电解液包含含氟化合物。
另外,本实施方式的金属化合物粒子群的制造方法包括:将作为基础的金属化合物粒子群与硅氧化物的原料混合的混合工序、以及将通过混合工序获得的粉末在300℃~600℃下加热的加热工序。
所述硅氧化物的原料可以相对于作为基础的金属化合物粒子群的重量为1wt%~20wt%的比例混合。
所述硅氧化物的原料可以相对于作为基础的金属化合物粒子群的重量为3wt%~20wt%的比例混合。
在所述混合工序中,可进一步混合一种或两种以上的碱性化合物。或者,也可在所述混合工序之前,包括利用碱性化合物对作为基础的金属化合物粒子群进行前处理的前处理工序。作为所述碱性化合物,可列举碱金属或碱土类金属的氢氧化物、乙酸盐、硫酸盐、碳酸盐、硝酸盐、氯化物、无机系碱性剂或有机系碱性剂。
[发明的效果]
根据本发明,可提供一种可抑制蓄电装置内部电阻增加的金属化合物粒子群、包含金属化合物粒子群的蓄电装置用电极、以及金属化合物粒子群的制造方法。
附图说明
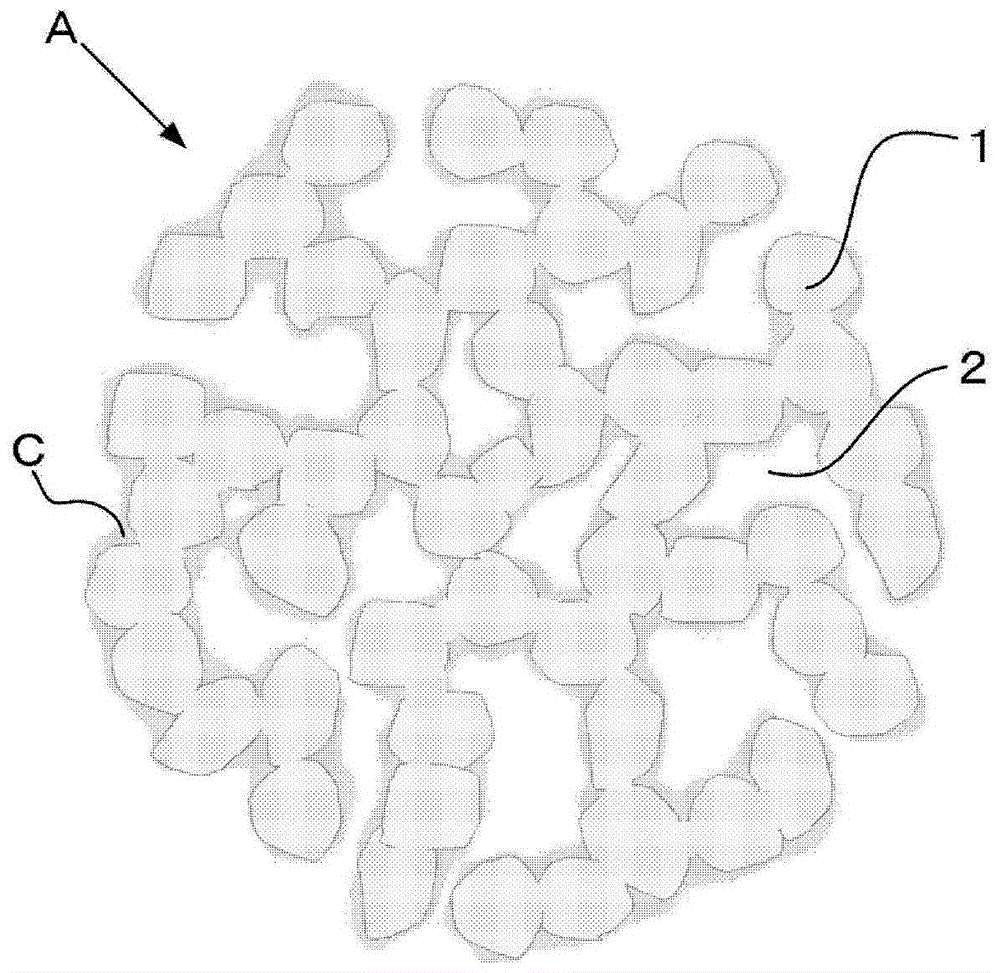
图1是表示本实施方式的金属化合物粒子群的概念图。
图2是比较例的钛酸锂粒子群的扫描电子显微镜(scanningelectronmicroscope,sem)图像,倍率为50.0k。
图3是钛酸锂粒子群的sem图像(×50.0k),(a)是比较例的钛酸锂粒子群的sem图像,(b)是实施例的钛酸锂粒子群的sem图像。
图4是说明对钛酸锂粒子群进行元素分析的结果的sem图像及图表,(a)是比较例的钛酸锂粒子群的结果,(b)是实施例的钛酸锂粒子群的结果。
图5是表示比较例的钛酸锂群的细孔径分布的图表。
图6是表示比较例及实施例的钛酸锂群的细孔径分布的图表。
图7是表示针对负极的活性物质层中包含使用具有丙烯酰基的硅烷偶联剂制作的比较例及实施例的钛酸锂粒子群的锂离子二次电池,测定初始的内部电阻的结果的图表。
图8是表示针对负极的活性物质层中包含使用具有丙烯酰基的硅烷偶联剂制作的和实施例的钛酸锂粒子群的锂离子二次电池,测定2000次循环后的内部电阻的增加率的结果的图表。
图9是表示针对负极的活性物质层中包含比较例与使用具有环氧基的硅烷偶联剂制作的实施例的钛酸锂粒子群的锂离子二次电池,测定初始的内部电阻的结果的图表。
图10是表示针对负极的活性物质层中包含比较例与使用具有环氧基的硅烷偶联剂制作的实施例的钛酸锂粒子群的锂离子二次电池,测定2000次循环后的内部电阻的增加率的结果的图表。
图11是表示针对负极的活性物质层中包含比较例与添加碱性化合物的同时而制作的实施例的钛酸锂粒子群的锂离子二次电池,测定了初始的内部电阻的结果的图表。
图12是表示针对负极的活性物质层中包含比较例与添加碱性化合物的同时制作的实施例的钛酸锂粒子群的锂离子二次电池,测定了2000次循环后的内部电阻的增加率的结果的图表。
图13是表示对比较例的钛酸锂粒子群进行热重量测定的结果的图表,(a)是热重量测定结果的图表,(b)是用于根据损失量换算硅烷偶联剂的量的标准曲线。
图14是表示硅烷偶联剂的添加量与钛酸锂粒子群中所含的si元素的量的关系的图表。
图15中的是表示进行实施例的钛酸锂粒子群的元素分析的结果的图表,(a)是表示碳浓度的图表,(b)是表示硅浓度的图表,(c)是表示碳/硅之比的图表。
图16是表示对加热处理前的钛酸锂粒子群及加热处理后的钛酸锂粒子群进行了x射线光电能谱分析的结果的图表。
图17是表示对加热处理后的钛酸锂粒子群进行了x射线衍射分析的结果的曲线图。
图18是表示对比较例与实施例的钛酸锂粒子群测定固体1h-核磁共振(nuclearmagneticresonance,nmr)的结果的图,(a)是表示固体1h-nmr的测定数据的图表,(b)是表示积分强度的运算结果的图表。
图19是表示测定使用比较例与实施例的钛酸锂粒子群制成的锂离子二次电池在2000次循环后的内部电阻的增加率的结果的图表。
具体实施方式
以下,对实施本发明的方式进行说明。再者,本发明不限于以下说明的实施方式。
[1.结构]
金属化合物粒子群主要用于蓄电装置的电极。作为构成金属化合物粒子群的金属化合物粒子,是能够作为锂离子二次电池或锂离子电容器等蓄电装置的正极活性物质或负极活性物质工作的材料。金属化合物粒子群具有纳米尺寸的金属化合物粒子相连而成的三维网状结构,在金属化合物粒子群的一部分上形成有包含硅氧化物(siox或siox(oh)4-2x(x为0~2))的涂布层。此包含硅氧化物的涂布层形成在金属化合物粒子的表面的至少一部分上。另外,在金属化合物粒子群的三维网状结构中存在空隙,在划定空隙的金属化合物粒子上,在其表面形成有包含硅氧化物的涂布层。
(金属化合物粒子群)
如图1所示,金属化合物粒子群a具有三维网状结构。三维网状结构表示纳米尺寸的金属化合物的一次粒子1键合而相连成网状,且存在纳米尺寸的空隙2的结构。一次粒子1的键合界面上不存在晶界,另一方面,一次粒子1之间存在大量的微小细孔。认为在所述金属化合物粒子群a中,三维网状结构形成负极活性物质的电子路径,空隙2成为电解液的储藏地,一次粒子1之间的细孔成为离子的路径。
金属化合物粒子是能够吸藏及放出锂离子的粒子。作为金属化合物粒子,可列举feo、fe2o3、fe3o4、mno、mno2、mn2o3、mn3o4、coo、co3o4、nio、ni2o3、tio、tio2、tio2(b)、tinb2o7、nb2o5、cuo、nio、sno、sno2、sio2、ruo2、wo、wo2、wo3、moo3、zno等氧化物;sn、si、al、zn等金属;livo2、li3vo4、li4ti5o12、sc2tio5、fe2tio5、li2na2ti6o14、li2bati6o14、li2srti6o14等复合氧化物;li2.6co0.4n、ge3n4、zn3n2、cu3n等氮化物;y2ti2o5s2、mos2。
这些金属化合物粒子中,特别对包含锂的氧化物或含氧酸盐进行具体说明。包含锂的氧化物或含氧酸盐利用liαmβyγ表示。在金属氧化物的情况下,例如m=co、ni、mn、ti、si、sn、al、zn、mg、nb、v中的任一种,y=o。在金属含氧酸盐的情况下,例如,m=fe、mn、v、co、ni中的任一种,y=po4、sio4、bo3、p2o7的任一种。mβ可为mδm'ε的合金,例如m=sn、sb、si的任一种,且m'=fe、co、mn、v、ti、ni的任一种。例如,可使用磷酸铁锂、钛酸锂、钴酸锂、磷酸钒锂、磷酸铁锰锂。其中,优选钛酸锂(li4ti5o12)。
三维网状结构的剖面中的空隙率优选为7%~50%的范围。若空隙率未满7%,则与电解液接触的面积少,会对电解液中的离子的移动造成影响。另外,如果空隙率超过50%,则一次粒子1彼此的键合变粗而难以形成三维网状结构。
一次粒子1的平均粒子径为5nm~300nm的范围。平均粒子径的算出方法如下所述。首先,使用扫描电子显微镜观察一次粒子1,拍摄至少含有150个以上一次粒子1的图像。对所拍摄的一个视野(图像)中所含的一次粒子1的椭圆形图像的长径与短径进行测定,针对各一次粒子1算出所述长径与短径的平均值。然后,将各一次粒子1的平均值相加,并且将其相加值除以所测定的一次粒子1的个数。其结果收敛于5nm~300nm的范围。
5~300范围的一次粒子1键合而形成三维网状结构时,金属化合物粒子会获得大量的纳米尺寸的细孔。如此,与电解液接触的金属化合物粒子的面积增加,电解液中的离子移动变得顺畅。另外,对所述金属化合物粒子的细孔进行测定,结果存在大量的微细的细孔。特别是,也包含大量的40nm以下的微细的细孔。
根据利用氮气吸附测定法对具有三维网状结构的金属化合物粒子进行测定而得的细孔分布而换算出差分细孔容积。其结果,10nm~40nm范围的细孔径中的差分细孔容积具有0.01cm3/g以上的值,特别是可获得大量0.02cm3/g以上的值。因此,与电解液接触的面积增大,与电解液接触的面积越多,用于负极时的放电速率特性越提高。
(涂布层)
涂布层c形成在金属化合物粒子群a的一部分上。特别是,可形成在金属化合物粒子表面的至少一部分上。涂布层c是包含硅氧化物的层。涂布层c也可形成在划定金属化合物粒子群a的空隙2的金属化合物粒子的表面。即,除了金属化合物粒子群a的外周面侧以外,在金属化合物粒子群a的内部结构中的空隙2中,在金属化合物粒子的表面也可形成涂布层c。但是,涂布层c未以填满空隙2的程度形成,在形成有涂布层c的金属化合物粒子群a中也残存有空隙2。此种金属化合物粒子群a具有对于10nm以下的细孔径而言为超过0.008dvp/drp的细孔容积变化率。另外,形成有包含硅氧化物的涂布层c的金属化合物粒子群a相对于未形成包含硅氧化物的涂布层c的金属化合物粒子群,积分强度的减少率为25%以上。
此种涂布层c优选形成为在金属化合物粒子群a中si元素的含量为0.4以上且3.0wt%以下,如果为此范围内,则可适当地抑制充电放电后的内部电阻的增加率。如果超过3.0wt%,则涂布层对抑制堆积物的产生是过剩的,无法期待进一步抑制内部电阻的增加率的效果,另一方面,涂布层的影响变大,初始的内部电阻增加。进而优选的是,涂布层c优选形成为在金属化合物粒子群a中si元素的含量为0.4以上且1.4wt%以下,如果为此范围内,则涂布层c平衡良好地形成在金属化合物粒子群a,初始的内部电阻值也受到抑制。
涂布层c中所含的硅氧化物具有低结晶性。即,硅氧化物为非晶,不具有完全的晶体结构。此种非晶状的硅氧化物被认为与金属化合物粒子的键合性良好。因此,包含非晶状的硅氧化物的涂布层c并非局部地形成在金属化合物粒子的表面。即,以均匀、且粘结在金属化合物粒子上的方式形成在金属化合物粒子的表面。虽然没有分析硅氧化物与金属化合物粒子的键合状态,但认为大概是金属化合物粒子的表面官能基与硅氧化物通过氢吸附等物理吸附的键合、或者是金属化合物粒子的表面官能基与硅氧化物的表面官能基的化学键合。
再者,涂布层c中除了硅氧化物以外还含有1%以下程度的碳。因此,推测涂布层c中含有碳化合物或siocnh2n+1或sic等成分。
所述的金属化合物粒子群a被用作混合电容器或锂离子二次电池等蓄电装置用的电极材料。以下,以使用本实施方式的金属化合物粒子群a的混合电容器的结构为一例进行说明。
(1-1.蓄电装置)
锂离子二次电池是蓄电装置的一例。锂离子二次电池中,使正极与负极隔着隔板相向而形成元件,并使电解液含浸于元件而成。锂离子二次电池根据正负极的锂离子的吸藏及放出方向进行充电放电。正极及负极分别将活性物质层与集电体一体化而成。正极活性物质与集电体、以及负极活性物质与集电体分别使用压接或刮刀法等经由粘合剂来接合。
典型而言,集电体为铝、铜、铁、镍、钛、钢、碳等导电材料。特别是,优选为具有高的热传导性与电子传导性的铝或铜。集电体的形状可采用膜状、箔状、板状、网状、多孔金属网状、圆筒状等任意形状。
作为粘合剂,例如可列举:氟系橡胶、二烯系橡胶、苯乙烯系橡胶等橡胶类,聚四氟乙烯、聚偏二氟乙烯等含氟聚合物,羧基甲基纤维素、硝基纤维素等纤维素,除此以外可列举聚烯烃树脂、聚酰亚胺树脂、丙烯酸树脂、腈树脂、聚酯树脂、酚树脂、聚乙酸乙烯酯树脂、聚乙烯醇树脂、环氧树脂等。这些粘合剂可单独使用,也可混合使用两种以上。
(1-2.正极)
正极活性物质层是将lmo(锰酸锂)粒子、作为导电助剂的碳及粘合剂混炼成型,与集电体合在一起。作为碳,只要具有导电性,就可并无特别限定地使用,例如可列举科琴黑、乙炔黑、槽法碳黑(channelblack)等碳黑、富勒烯、碳纳米管、碳纳米纤维、无定形碳、碳纤维、天然石墨、人造石墨、石墨化科琴黑、介孔碳、气相法碳纤维等。作为粘合剂,使用聚四氟乙烯、聚偏二氟乙烯(polyvinylidenefluoride,pvdf)、四氟乙烯-六氟丙烯共聚物、聚氟乙烯、羧甲基纤维素等公知的粘合剂。粘合剂的含量相对于电极材料的总量优选为1以上且30质量%以下。
在所述正极活性物质层中,也可以添加其他正极活性物质。作为其他正极活性物质,可列举层状岩盐型limo2、层状li2mno3-limo2固溶体、以及尖晶石型lim2o4(式中的m是指fe、co、ni、它们的组合、或者它们与mn的组合)。作为它们的具体例,可列举licoo2、linio2、lini4/5co1/5o2、lini1/3co1/3mn1/3o2、lini1/2mn1/2o2、lifeo2、limno2、li2mno3-licoo2、li2mno3-linio2、li2mno3-lini1/3co1/3mn1/3o2、li2mno3-lini1/2mn1/2o2、li2mno3-lini1/2mn1/2o2-lini1/3co1/3mn1/3o2、limn3/2ni1/2o4。
作为所述以外的其他正极活性物质,除了硫及li2s、tis2、mos2、fes2、vs2、cr1/2v1/2s2等硫化物、nbse3、vse2、nbse3等硒化物、cr2o5、cr3o8、vo2、v3o8、v2o5、v6o13等氧化物之外,可列举lini0.8co0.15al0.05o2、livopo4、liv3o5、liv3o8、mov2o8、li2fesio4、li2mnsio4、lifepo4、life1/2mn1/2po4、limnpo4、li3v2(po4)3等复合氧化物。
(1-3.负极)
负极活性物质为所述金属化合物粒子群a,且是能够吸藏及放出锂离子的金属化合物粒子群。例如,钛酸锂具有利用锂离子的插入·脱离的能量储存功能。所述插入·脱离的体积变化为约1%,因此电容劣化少。充电放电电位为约1.55v(vs.li/li+),因此不易产生因电解液的分解或极速充电放电而引起锂金属的析出等副反应,且循环特性优异。
(1-4.隔板)
作为成为基材的隔板,可将牛皮纸、马尼拉麻、针茅纤维、大麻、人造丝等纤维素及这些的混合纸,聚对苯二甲酸乙二酯、聚对苯二甲酸丁二酯、聚萘二甲酸乙二酯、这些的衍生物等聚酯系树脂,聚四氟乙烯系树脂、聚偏二氟乙烯系树脂、维尼龙系树脂、脂肪族聚酰胺、半芳香族聚酰胺、全芳香族聚酰胺等聚酰胺系树脂,聚酰亚胺系树脂、聚乙烯树脂、聚丙烯树脂、三甲基戊烯树脂、聚苯硫醚树脂、丙烯酸树脂等树脂单独使用或混合使用。
(1-5.电解液)
电解液的电解质是成为锂离子源的锂盐。锂盐为lipf6、libf4、liclo4、lin(so2cf3)2、lin(so2c2f5)2、cf3so3li、lic(so2cf3)3及lipf3(c2f5)3或这些的混合物。锂盐的浓度一般在0.1mol/l~2.5mol/l的范围,优选在0.5mol/l~2mol/l的范围。
作为电解质,除锂盐以外,也可添加四级铵盐。相对于锂盐为1.6m以上的摩尔浓度而言,四级铵盐并无特别限定,例如以1m的摩尔浓度添加。作为四级铵盐,阳离子可列举四乙基铵、三乙基甲基铵、二乙基二甲基铵、乙基三甲基铵、甲基乙基吡咯烷鎓、螺环联吡咯烷鎓、螺环-(n,n')-联吡咯烷鎓、1-乙基-3-甲基咪唑鎓、1-乙基-2,3-二甲基咪唑鎓等,阴离子可列举bf4-、pf6-、clo4-、asf6-、sbf6-、alcl4-或rfso3-、(rfso2)2n-、rfco2-(rf为碳数1~8的氟烷基)等。
作为电解液的溶媒,可使用以下所列举者。再者,这些溶媒可分别单独使用,也可混合使用两种以上。例如,可列举:环状碳酸酯、链状碳酸酯、磷酸酯、环状醚、链状醚、内酯化合物、链状酯、腈化合物、酰胺化合物、砜化合物等。作为环状碳酸酯,可列举碳酸亚乙酯、碳酸亚丙酯、碳酸亚丁酯、4-氟-1,3-二氧杂环戊烷-2-酮、4-(三氟甲基)-1,3-二氧杂环戊烷-2-酮等,优选为碳酸亚乙酯、碳酸亚丙酯。
作为链状碳酸酯,可列举碳酸二甲酯、碳酸乙基甲酯、碳酸二乙酯、碳酸甲基正丙酯、碳酸甲基异丙酯、碳酸正丁基甲酯、碳酸二乙酯、碳酸乙基正丙酯、碳酸乙基异丙酯、碳酸正丁基乙酯、碳酸二正丙酯、碳酸二异丙酯、碳酸二正丁酯、碳酸氟乙基甲酯、碳酸二氟乙基甲酯、碳酸三氟乙基甲酯等,优选为碳酸二甲酯、碳酸乙基甲酯、碳酸二乙酯。
作为磷酸酯,可列举:磷酸三甲酯、磷酸三乙酯、磷酸乙基二甲酯、磷酸二乙基甲酯等。作为环状醚,可列举四氢呋喃、2-甲基四氢呋喃等。作为链状醚,可列举二甲氧基乙烷等。作为内酯化合物,可列举γ-戊内酯、γ-丁内酯等。作为链状酯,可列举:丙酸甲酯、乙酸甲酯、乙酸乙酯、甲酸甲酯等。作为腈化合物,可列举乙腈等。作为酰胺化合物,可列举二甲基甲酰胺等。作为砜化合物,可列举环丁砜、甲基环丁砜、二甲基砜、乙基甲基砜、异丙基砜等,但并不限定于这些。
[2.制造方法]
本发明的金属化合物粒子群的制造方法具有以下工序。
(1)混合作为基础的金属化合物粒子群与硅氧化物的原料的混合工序
(2)将通过混合工序获得的粉末在300℃~600℃下加热的加热工序
再者,本实施方式的金属化合物粒子群a是基于作为基础的金属化合物粒子群而制成。作为基础的金属化合物粒子群具有三维网状结构。但是,金属化合物粒子群的表面未形成涂布层。通过使用所述金属化合物粒子群进行所述(1)及(2)的工序中记载的处理,制造本实施方式的金属化合物粒子群a。以下,详细说明各工序。
(1)混合工序
在混合工序中,将作为基础的金属化合物粒子群与硅氧化物的原料混合,制作混合溶液。作为硅氧化物的原料,可使用包含硅原子的硅烷偶联剂。具体地说,在混合作为基础的金属化合物粒子群与硅烷偶联剂而得者中添加溶媒,制成混合溶液。
硅烷偶联剂优选以相对于作为基础的金属化合物粒子群的重量为1wt%~20wt%的比例混合,更优选以3wt%~20wt%的比例混合。硅烷偶联剂的混合量小于1wt%时,在使用金属化合物粒子群a制成的蓄电装置中,负极材料上会明显生成堆积物。因此,无法获得抑制内部电阻的效果。另外,硅烷偶联剂的混合量超过20wt%时,在使用金属化合物粒子群a制成的蓄电装置中,形成的涂布层c的厚度增加。
因此,如果硅烷偶联剂的混合量为3wt%~20wt%,则通过加热工序,形成金属化合物粒子群a中含有的si元素的量为0.4以上且3.0wt%以下的涂布层c,可适当地抑制充电放电后的内部电阻的增加率。另外,通过使硅烷偶联剂的混合量为3wt%~10wt%,经过加热工序,形成金属化合物粒子群a中含有的si元素的量为0.4以上且1.4wt%以下的涂布层c,涂布层c的厚度变得适当,可适当地抑制充电放电后的内部电阻的增加率,同时抑制蓄电装置的初始的内部电阻增加。
硅烷偶联剂并无特别限定,代表性的是下述通式所表示的化合物或包含它们的盐的硅烷偶联剂。
[化1]
(式中,x1为具有乙烯基、氨基、环氧基、氯基、巯基、酮亚胺基、丙烯酰基、甲基丙烯酰基、苯乙烯基、苯基、异氰酸酯基、烷基、烷氧基的原子团、x2~x4为烷基、烷氧基、氯基、羟基)
作为这些硅烷偶联剂,可例示四乙氧基硅烷、乙烯基三甲氧基硅烷、n-(2-氨基乙基)3-氨基丙基三甲氧基硅烷、3-氨基丙基三乙氧基硅烷、3-缩水甘油氧基丙基三甲氧基硅烷、2-(3,4-环氧基环己基)乙基三甲氧基硅烷、3-丙烯酰氧基丙基三甲氧基硅烷、3-甲基丙烯酰氧基丙基三甲氧基硅烷、3-巯基丙基三甲氧基硅烷、n-〔2-(乙烯基苄基氨基)乙基〕-3-氨基丙基三甲氧基硅烷-盐酸盐等。
作为溶媒,只要是对反应没有不良影响的液体就可并无特别限定地使用,可适当使用水、甲醇、乙醇、异丙醇等。也可混合两种以上的溶媒使用。
所制成的混合溶液使用均化器(homogenizer)等充分搅拌,以使金属化合物粒子群与硅烷偶联剂分散在混合溶液中。通过此搅拌,成为硅烷偶联剂附着在作为基础的金属化合物粒子群的表面及空隙的状态。静置搅拌后的混合溶液,使分散在混合溶液中的金属化合物粒子群与硅烷偶联剂凝聚,而回收金属化合物粒子群与硅烷偶联剂的混合粉末。
此处,混合溶液中可进一步添加碱性化合物。通过添加碱性化合物,相对于制造过程中投入的硅氧化物的原料的量,硅氧化物的附着量增多,可以少的硅烷偶联剂的投入量,更好地抑制充电放电后的内部电阻的增加率。或者在想要获得与未添加碱性化合物时相同的抑制效果的情况下,可减少制造过程中投入的硅氧化物的原料的量,从而导致成本的减少。只是推测,并不限于此,但可认为通过碱性化合物的催化作用,金属化合物粒子群与硅烷偶联剂的反应性提高,硅氧化物容易附着在金属化合物粒子群的表面。因此,可认为,相对于制造过程中投入的硅氧化物的原料的量,硅氧化物的附着量变多,与不添加碱性化合物的情况相比,充电放电后的内部电阻的增加率得到了抑制。再者,碱性化合物的添加量相对于10g金属化合物粒子群优选为0.05mmol~5mmol的范围。
作为能够添加到混合溶液中的碱性化合物,可列举碱金属或碱土类金属的氢氧化物、乙酸盐、硫酸盐、碳酸盐、硝酸盐、氯化物、无机系碱性剂或有机系碱性剂。碱金属及碱土类金属例如为锂、钠、钾及钙等。无机系碱性剂例如可列举氨水。作为有机系碱性剂,可列举三乙胺、二乙胺、吡啶、四甲基胍等胺类。这些碱性化合物可添加一种或两种以上到混合溶液中。
再者,作为优选添加的碱性化合物,如锂盐那样,是与金属化合物粒子群的原料或电解液中的电解质相同的种类,在金属化合物粒子群的特性中不成为杂质,或者通过制造过程中的热处理等被除去,不残存在金属化合物粒子群中。
另外,除了在混合溶液中进一步添加碱性化合物的方法以外,也可利用碱性化合物对金属化合物粒子群进行前处理,然后转移到与硅烷偶联剂的混合工序。在前处理工序中,将碱性化合物及金属化合物粒子群与溶媒一起混合,并进行过滤干燥。然后,使用经过滤及干燥者,进入混合工序。混合后,可以30℃~100℃的范围放置1~100小时。如此种方法这样,预先利用碱性化合物对金属化合物粒子群进行前处理,可更好地抑制充电放电后的内部电阻的增加率。再者,此方法中的溶媒及碱性化合物可使用与在混合溶液中进一步添加碱性化合物的方法相同的种类。
(2)加热工序
在加热工序中,对混合工序中获得的混合粉末进行加热处理。加热处理以300℃~600℃的温度范围进行1~24小时即可。认为在此加热工序中,源自硅烷偶联剂的硅氧化物以非晶的状态生成,与作为基础的金属化合物粒子群键合。
再者,也可在加热工序之前,回收混合工序中获得的凝聚物,对凝聚物实施干燥处理。干燥优选设为加热干燥,以20℃~180℃加热凝聚物使其干燥。另外,加热时间设为6~48小时即可。通过此干燥工序,可获得在凝聚物中含有的溶媒通过加热而蒸发的状态下,混合了作为基础的金属化合物粒子群与硅烷偶联剂而得的混合粉末。
[3.作用效果]
本实施方式的金属化合物粒子群a及含有此金属化合物粒子群a而构成的蓄电装置用电极发挥的作用效果如下。
(1)一种金属化合物粒子群,具有金属化合物粒子相连而成的三维网状结构,在此金属化合物粒子群的一部分上形成有包含硅氧化物的涂布层。
在金属化合物粒子群a用作蓄电装置用电极的负极活性物质的情况下,利用涂布层c防止在负极上产生堆积物。因此,可抑制蓄电装置的内部电阻的增加。
(2)包含硅氧化物的涂布层c形成在金属化合物粒子的表面的至少一部分上。
通过在金属化合物粒子的表面形成涂布层c,更可靠地防止在负极表面产生堆积物。因此,可抑制蓄电装置的内部电阻的增加。
(3)基于硅氧化物含有的si元素的量相对于金属化合物粒子群a整体为0.4以上且3.0wt%以下。
通过以基于硅氧化物含有的si元素的量相对于金属化合物粒子群a整体为0.4以上且3.0wt%以下的方式形成涂布层c,可赋予涂布层c适当的厚度,抑制蓄电装置充电放电后的内部电阻的增加率。
(4)基于硅氧化物含有的si元素的量相对于金属化合物粒子群整体为0.4以上且1.7wt%以下。
通过以基于硅氧化物含有的si元素的量相对于金属化合物粒子群a整体为0.4以上且1.7wt%以下的方式形成涂布层c,可进一步赋予涂布层c适当的厚度,抑制蓄电装置充电放电后的内部电阻的增加率,并且还可抑制蓄电装置的初始的内部电阻的增加。
(5)三维网状结构中存在空隙2,在划定空隙2的金属化合物粒子上,在其表面形成有包含硅氧化物的涂布层c。
涂布层c不仅形成在金属化合物粒子a的外周面,在金属化合物粒子群a的内部结构中的空隙2中也形成在金属化合物粒子的表面。通过使涂布层c以到达空隙的程度充分地形成,可提高分别抑制堆积物的产生及内部电阻的增加的效果。
(6)涂布层c中含有的硅氧化物为非晶状的硅氧化物。
通过在涂布层c中包含非晶状的硅氧化物,提高了涂布层c与金属化合物粒子的键合性。因此,可获得涂布层c以均匀且粘结在金属化合物粒子上的方式键合的金属化合物粒子群a。在将此种金属化合物粒子群a用于蓄电装置的情况下,涂布层c不会被电解液等溶解。因此,在蓄电装置中,能够可靠地抑制堆积物的产生及内部电阻的增加。
(7)具有根据利用氮气吸附测定法测定金属化合物粒子群而得的细孔分布换算的、相对于10nm以下的细孔径而言为超过0.008dvp/drp的细孔容积变化率。
在具有相对于10nm以下的细孔径而言为超过0.008dvp/drp的细孔容积变化率的金属化合物粒子群中,消除了空隙的堵塞。即,涂布层c未以填埋空隙的方式形成。因此,能够抑制初始的内部电阻的增加。
(8)相对于未形成包含硅氧化物的涂布层的金属化合物粒子群,积分强度的减少率为25%以上。
相对于未被覆包含硅氧化物的涂布层的金属化合物粒子群,在形成有涂布层c的金属化合物粒子群中,积分强度比降低。在此种本实施方式的金属化合物粒子群中,利用涂布层c抑制负极上的堆积物生成,因此可抑制内部电阻的增加。
另外,作为蓄电装置,可发挥以下的作用效果。
(9)包括:具有包含所述金属化合物粒子群a而构成的负极、及正极的元件、以及含浸在元件中的电解液,且电解液包含含氟化合物。
认为在蓄电装置中,例如在电解液中使用作为含氟化合物的lipf6或libf4等情况下,电解液中的成分在负极表面发生反应,而在负极上产生来自氟的堆积物。推测本实施方式的金属化合物粒子群a特别是对抑制这些来自氟的堆积物生成有效果。
另外,作为金属化合物粒子群的制造方法,可起到以下的作用效果。
(10)包括:混合作为基础的金属化合物粒子群与硅氧化物的原料的混合工序、及将通过混合工序获得的粉末在300℃~600℃下加热的加热工序。
通过在300℃~600℃下对作为基础的金属化合物粒子群与硅氧化物的原料的混合粉末进行加热处理,在金属化合物粒子的表面形成包含来自硅氧化物的原料的硅氧化物的涂布层c。通过此加热处理,硅氧化物成为非晶状态,与金属化合物粒子的键合得到提高。
(11)硅氧化物的原料以相对于作为基础的金属化合物粒子群的重量为1wt%~20wt%的比例混合。
通过混合1wt%~20wt%的硅氧化物的原料,在金属化合物粒子上形成适当量的涂布层c。即,能够形成可抑制初始的内部电阻的增加、同时抑制充电放电后的内部电阻的增加的涂布层c。
(12)硅氧化物的原料以相对于作为基础的金属化合物粒子群的重量为3wt%~20wt%的比例混合。
通过混合3wt%~20wt%的硅氧化物的原料,利用在300℃~600℃下的加热处理,形成金属化合物粒子群a中含有的si元素的量为0.4以上且3.0wt%以下的适当厚度的涂布层c,可适当抑制充电放电后的内部电阻的增加率。
(13)硅氧化物的原料以相对于作为基础的金属化合物粒子群的重量为3wt%~10wt%的比例混合。
通过混合3wt%~10wt%的硅氧化物的原料,利用在300℃~600℃下的加热处理,形成金属化合物粒子群a中含有的si元素的量为0.4以上且1.7wt%以下的更适当厚度的涂布层c,可适当抑制充电放电后的内部电阻的增加率,并且抑制蓄电装置的初始的内部电阻的增加。
(14)在混合工序中,进一步混合一种或两种以上的碱性化合物。碱性化合物例如可列举碱金属或碱土类金属的氢氧化物、乙酸盐、硫酸盐、碳酸盐、硝酸盐、氯化物、无机系碱性剂或有机系碱性剂。
如果在混合工序中添加所述碱性化合物,则相对于制造过程中投入的硅氧化物的原料的量,硅氧化物的附着量变多,可以少的硅烷偶联剂的投入量,更好地抑制充电放电后的内部电阻的增加率。或者在想要获得与未添加碱性化合物的情况相同的抑制效果的情况下,可减少制造过程中投入的硅氧化物的原料的量,从而导致成本的减少。
(15)或者在混合工序之前,利用碱性化合物对作为基础的金属化合物粒子群进行前处理。
如果预先利用碱性化合物对金属化合物粒子群进行前处理,进而可更好地抑制充电放电后的内部电阻的增加率。
[实施例]
使用以下的实施例对本发明进行说明,但本发明不限于以下的实施例。
(实施例1)
实施例1中,使用钛酸锂作为成为基础的金属化合物粒子群、使用含有丙烯酰基作为有机官能基的(ch3o)3sic3h6ococh=ch2作为硅烷偶联剂。硅烷偶联剂相对于钛酸锂混合了5wt%。在混合工序中,使用均化器以转速15000搅拌10分钟。回收静置后的凝聚物,在150℃下真空干燥12小时。然后,对干燥后的粉末在400℃下进行3小时加热处理。
(实施例2)
在实施例2中,相对于钛酸锂混合了10wt%的硅烷偶联剂。除此之外,与实施例1同样地制作。
(实施例3)
在实施例3中,相对于钛酸锂混合了20wt%的硅烷偶联剂。除此之外,与实施例1同样地制作。
(实施例4)
在实施例4中,相对于钛酸锂混合了3wt%的硅烷偶联剂。除此之外,与实施例1同样地制作。
(实施例5)
在实施例5中,作为硅烷偶联剂,使用含有环氧基作为有机官能基的3-缩水甘油氧基丙基三甲氧基硅烷,相对于钛酸锂混合了3wt%。除此之外,与实施例1同样地制作。即,与实施例4相比,不同点在于硅烷偶联剂为环氧系。
(实施例6)
在实施例6中,作为硅烷偶联剂使用3-缩水甘油氧基丙基三甲氧基硅烷,相对于钛酸锂混合了5wt%。除此之外,与实施例1同样地制作。即,与实施例5同样,硅烷偶联剂为环氧系,但与实施例5相比,硅烷偶联剂的添加量不同。
(实施例7)
在实施例7中,作为硅烷偶联剂使用3-缩水甘油氧基丙基三甲氧基硅烷,相对于钛酸锂混合了10wt%。除此之外,与实施例1同样地制作。即,与实施例5同样,硅烷偶联剂为环氧系,但与实施例5相比,硅烷偶联剂的添加量不同。
(实施例8)
在实施例8中,作为硅烷偶联剂使用3-缩水甘油氧基丙基三甲氧基硅烷,相对于钛酸锂混合了20wt%。除此之外,与实施例1同样地制作。即,与实施例5同样,硅烷偶联剂为环氧系,但与实施例5相比,硅烷偶联剂的添加量不同。
(实施例9)
在实施例9中,作为硅烷偶联剂使用3-缩水甘油氧基丙基三甲氧基硅烷,相对于钛酸锂混合了3wt%。在实施例9中,进而,除了添加钛酸锂与硅烷偶联剂之外,还添加了作为碱性化合物的氢氧化锂,经过了混合工序。相对于10g钛酸锂,以0.25mmol的比例添加氢氧化锂。除此之外,与实施例1同样地制作。即,与实施例5相比,不同点在于添加了碱性化合物。
(实施例10)
在实施例10中,作为硅烷偶联剂使用3-缩水甘油氧基丙基三甲氧基硅烷,相对于钛酸锂混合了5wt%。除此之外,与实施例9同样地制作。即,与实施例9同样地添加有碱性化合物,但与实施例9相比,硅烷偶联剂的添加量不同。
(实施例11)
在实施例11中,作为硅烷偶联剂使用3-缩水甘油氧基丙基三甲氧基硅烷,相对于钛酸锂混合了10wt%。除此之外,与实施例9同样地制作。即,与实施例9同样地添加有碱性化合物,但与实施例9相比,硅烷偶联剂的添加量不同。
(实施例12)
在实施例12中,作为硅烷偶联剂使用3-缩水甘油氧基丙基三甲氧基硅烷,相对于钛酸锂混合了20wt%。除此之外,与实施例9同样地制作。即,与实施例9同样地添加有碱性化合物,但与实施例9相比,硅烷偶联剂的添加量不同。
(实施例13)
在实施例13中,作为硅烷偶联剂使用3-缩水甘油氧基丙基三甲氧基硅烷,相对于钛酸锂混合了3wt%。但是,在添加硅烷偶联剂的混合工序之前,将作为碱性化合物的氢氧化锂、作为溶媒的水、及作为基础的金属化合物粒子群的钛酸锂混合,利用搅拌器搅拌30分钟,在60℃下放置64小时后,过滤、水洗后,进行在30℃下干燥24小时的前处理。相对于10g钛酸锂,以2.5mmol的比例添加氢氧化锂。除此之外,与实施例9同样地制作。即,与实施例9相比,不同点在于,作为基础的金属化合物粒子群利用碱性化合物进行前处理。
(比较例1)
在比较例1中,不混合硅烷偶联剂,对钛酸锂进行了与实施例1同样的处理。
(比较例2)
在比较例2中,没有混合硅烷偶联剂。另外,混合工序后只进行真空干燥,不进行加热处理。
(比较例3)
在比较例3中,相对于钛酸锂混合了5wt%的硅烷偶联剂。另外,混合工序后只进行真空干燥,不进行加热处理。
(比较例4)
在比较例4中,相对于钛酸锂混合了10wt%的硅烷偶联剂。另外,混合工序后只进行真空干燥,不进行加热处理。
(比较例5)
在比较例5中,相对于钛酸锂混合了20wt%的硅烷偶联剂。另外,混合工序后只进行真空干燥,不进行加热处理。
(比较例6)
在比较例6中,相对于钛酸锂混合了3wt%的硅烷偶联剂。另外,混合工序后只进行真空干燥,不进行加热处理。
(关于涂布层的形成)
首先,以下说明对获得的钛酸锂粒子群进行观察的结果。图2~图4是利用扫描型电子显微镜对比较例1、比较例4及实施例2的钛酸锂粒子群进行拍摄而得的sem。
图2是比较例1的钛酸锂粒子群的sem图像(×50.0k)。未添加硅烷偶联剂的比较例1的钛酸锂粒子群具有三维网状结构。但是,在钛酸锂粒子群的外周面及空隙的表面未形成涂布层c。
图3是比较例4与实施例2的钛酸锂粒子群的sem图像。图3(a)是比较例4的钛酸锂粒子群的sem图像,倍率上段为×50.0k,下段为×100k。由图3(a)也明确可知,在混合10wt%的硅烷偶联剂而仅进行干燥的比较例4中,钛酸锂粒子群的外周面及空隙的表面为被硅烷偶联剂覆盖的状态。硅烷偶联剂以填埋钛酸锂粒子群的空隙的方式附着在钛酸锂粒子群上。
另一方面,图3(b)是实施例2的钛酸锂粒子群的sem图像,倍率上段为×50.0k、下段为×100k。由图3(b)也明确可知,在混合10wt%的硅烷偶联剂并在干燥之后进行加热的实施例2中,在钛酸锂粒子群的外周面及空隙的表面形成有包含来自硅烷偶联剂的硅氧化物的涂布层。涂布层在钛酸锂粒子群的空隙中也形成于钛酸锂粒子的表面,但不是以填埋空隙的方式形成的状态。
此处,图4中示出对比较例4与实施例2的钛酸锂粒子群进行元素分析的结果。图4(a)表示比较例4的钛酸锂粒子群的分析结果,图4(b)表示实施例2的钛酸锂粒子群的分析结果。元素分析如各图中的sem图像中箭头所示,以横穿钛酸锂粒子群的直径部分的方式进行。另外,在图中的图表中,纵轴描绘si/ti比,验证了含有来自硅烷偶联剂的si的比例。
由图4(a)及(b)可确认,在比较例4与实施例2的钛酸锂粒子组中,不仅在钛酸锂粒子群的外表面,而且在内部结构的空隙部分的粒子表面也存在si。即,可知在比较例4与实施例2的钛酸锂粒子群双方中,并非局部性而是整体上存在si。
(关于细孔径分布)
接着,对所获得的实施例1及实施例2、比较例2~比较例4的钛酸锂粒子群测定细孔径分布。作为测定方法,使用氮气吸附测定法。具体而言,向钛酸锂粒子表面及与钛酸锂粒子表面连通的内部形成的细孔导入氮气,求出氮气的吸附量。接着,使所导入的氮气的压力缓缓增加,描绘氮气相对于各平衡压的吸附量,从而获得吸附等温曲线。测定使用高精度气体/蒸气吸附量测定装置拜尔索尔浦(belsorp)-max-n(日本拜尔(bel)股份有限公司制造)进行。
图5及图6是横轴取细孔径、纵轴描绘细孔容积变化率dvp/d(rp)的图表。图5表示未进行加热的比较例2~比较例4的细孔径分布数据。由图5明确可知,相对于未混合硅烷偶联剂的比较例2,混合了硅烷偶联剂的比较例3及比较例4中,10nm以下的空隙显著减少。即,可知在混合了硅烷偶联剂的比较例3及比较例4中,空隙被硅烷偶联剂填充。
图6表示进行了加热的、比较例1、实施例1、及实施例2的细孔径分布数据。由图6明确可知,混合了硅烷偶联剂的实施例1及实施例2具有与未混合硅烷偶联剂的比较例1相同程度的空隙。具体地说,在实施例1及实施例2中,关于10nm以下细孔径的细孔容积变化率,具有超过0.008dvp/drp的值。再者,细孔容积变化率在10nm以下的细孔径中不需要一律超过0.008,一部分超过0.008即可。可知,在混合硅烷偶联剂进行加热的实施例1及实施例2中,通过形成包含来自硅烷偶联剂的硅氧化物的涂布层,消除了空隙的堵塞。
(关于内部电阻)
为了验证内部电阻,使用实施例1~实施例4、比较例1~比较例6的钛酸锂粒子群制作锂离子二次电池。具体地说,相对于各粒子群,加入5重量%的聚偏二氟乙烯及适量的n-甲基吡咯烷酮充分混炼,形成浆料,涂布在铝箔上,并进行干燥而获得负极。另外,将lmo粒子、作为导电助剂的乙炔黑、及作为粘合剂的聚偏二氟乙烯(polyvinylidenefluoride,pvdf)添加到溶媒中,利用均化器进行搅拌,而生成浆料状的混合溶液。将此混合溶液涂布在铝集电箔上并干燥、进行冲压,将由此获得的正极活性物质层的片材作为正极。使纤维素的隔板介于前面的正极电极与负极电极之间而形成元件,并使元件含浸电解液。电解液使用1.6摩尔浓度的libf4作为溶质,使用γ-丁内酯(gamma-butyrolactone,gbl)作为溶媒。然后,使用层压膜对含浸有电解液的元件进行热封,制作了实施例1~实施例4、比较例1~比较例6的锂离子二次电池。
图7中示出对如此制成的实施例1~实施例4及比较例1~比较例6的锂离子二次电池测定初始的内部电阻(直流内部电阻(directcurrentinternalresistance,dcir))的结果。在图7中,连接圆形标记的点的系列是经过加热工序的系列,根据比较例1及实施例1~实施例4的结果而成,连接三角标记的点的系列是省略了加热工序的系列,根据比较例2~比较例6的结果而成。
根据图7,未混合硅烷偶联剂的比较例1及比较例2中,初始的dcir未发生变化。接着,参照圆形标记的系列,比较进行了加热处理的比较例1与实施例1~实施例4,混合3wt%硅烷偶联剂的实施例4、混合5wt%硅烷偶联剂的实施例1、混合10wt%硅烷偶联剂的实施例2与比较例1相比,内部电阻虽然增加,但增加倾向缓慢。另一方面,研究混合20wt%硅烷偶联剂的实施例3时,与比较例1相比,内部电阻大幅增加。因此,认为混合超过20wt%硅烷偶联剂时,内部电阻会进一步增加。
另外,参照三角标记的系列对未进行加热处理的比较例2~比较例6进行研究时,混合了硅烷偶联剂的比较例3~比较例6与未混合硅烷偶联剂的比较例2相比,内部电阻大幅增加。比较三角标记的系列与圆形标记的系列可知,在各混合比例中,进行了加热处理的实施例1~实施例4的内部电阻的增加得到抑制。认为这是因为在未进行加热处理的比较例3~比较例6中,硅烷偶联剂填埋了空隙,因此电解液的含浸性降低,初始的内部电阻增加。另一方面,在进行了加热处理的实施例1~实施例4中,涂布层未以填埋空隙的方式形成。因此,认为初始的内部电阻的增加受到了抑制。推测可能是硅烷偶联剂的有机部位因加热处理而烧毁,形成了空隙所致。
接着,在60℃下,进行电压范围为1.5以上且2.7v以下、电流为20ma的充电放电试验,由初始的内部电阻与充电放电后的内部电阻求出内部电阻的增加率。图8表示2000次循环后的内部电阻的增加率。内部电阻的增加率由充电放电后的内部电阻/初始的内部电阻求出。在图8中,连接圆形标记的点的系列是经过加热工序的系列,根据比较例1及实施例1~实施例4的结果而成,连接三角标记的点的系列是省略了加热工序的系列,根据比较例2~比较例6的结果而成。
首先,在比较例1及比较例2中,内部电阻的增加率均为166.4%。在比较例1及比较例2中,未混合硅烷偶联剂,因此在负极上产生了堆积物,内部电阻显著增加。参照圆形标记的系列,将进行了加热处理的比较例1与实施例1~实施例4进行比较时,首先,在混合了3wt%的硅烷偶联剂的实施例4中,增加率为118.4%,与比较例1相比,增加率得到抑制。另外,在混合了5wt%硅烷偶联剂的实施例1中,增加率为93.3%,与比较例1相比,增加率得到抑制。另外,混合10wt%硅烷偶联剂的实施例2的增加率为61.2%,提高了抑制增加率的效果。接着,混合20wt%硅烷偶联剂的实施例3的增加率为62.5%,比实施例2略有上升。根据此结果可知,在混合10wt%以上的硅烷偶联剂的情况下,增加率的抑制效果也变得平稳。
由以上内容可知,混合超过20wt%的硅烷偶联剂时,初始的内部电阻显著增加。另外,可知只要混合1wt%以上的硅烷偶联剂,就可防止堆积物的生成,抑制充电放电后的内部电阻的增加。特别是明确了通过将硅烷偶联剂的混合量设为3wt%~10wt%,可抑制蓄电装置初始的内部电阻的增加,并且可适当地抑制充电放电后的内部电阻的增加率。
另外,参照三角标记的系列对未进行加热处理的比较例2~比较例6进行研究时,混合了硅烷偶联剂的比较例3~比较例6与未混合硅烷偶联剂的比较例2相比,内部电阻的增加率减少。因此,确认了通过混合硅烷偶联剂可获得抑制增加率的效果。
但是,如果比较三角标记的系列与圆形标记的系列,则可知在各混合比例下,进行了加热处理的实施例1~实施例4的内部电阻的增加率较低。认为这是因为,在未进行加热处理的比较例3~比较例6中,硅烷偶联剂未在钛酸锂粒子群的表面形成涂布层,硅烷偶联剂仅附着在钛酸锂粒子群的表面。因此,在比较例3~比较例6中,发现了附着在钛酸锂粒子群表面的硅烷偶联剂被电解液溶解,在负极上生成了堆积物的可能性。
进而,制作了将使用环氧系的硅烷偶联剂制作的实施例5~实施例8的钛酸锂粒子群作为负极的活性物质层的锂离子二次电池。这些锂离子二次电池的制造方法及制造条件与使用实施例1~实施例4、比较例1~比较例6的钛酸锂粒子群的锂离子二次电池相同。然后,与使用未添加硅烷偶联剂的比较例1的钛酸锂粒子群的锂离子二次电池一起,测定了使用实施例5~实施例8的钛酸锂粒子群的锂离子二次电池的初始的内部电阻(dcir)以及2000次循环后的内部电阻的增加率。初始的内部电阻及内部电阻的增加率的测定方法与实施例1~实施例4及比较例1相同。
图9及图10中示出所述测定结果。图9是表示硅烷偶联剂的添加量与初始的内部电阻(dcir)的关系的图表。图10是表示硅烷偶联剂的添加量与内部电阻的增加率的关系的图表。如图9及图10所示,确认了即使改变硅烷偶联剂的种类,初始的内部电阻(dcir)及内部电阻的增加率也显示出与实施例1~实施例4相同的倾向。
即,确认了硅烷偶联剂与种类无关,只要混合1wt%以上,就会防止生成堆积物。而且,确认了如果硅烷偶联剂的添加量为20wt%以下,则与硅烷偶联剂的种类无关,随着硅烷偶联剂的增加的内部电阻的增加会缓慢收敛。进而,明确了通过将硅烷偶联剂的混合量设为3wt%~10wt%,则与硅烷偶联剂的种类无关,可抑制蓄电装置初始的内部电阻的增加,并且适当地抑制充电放电后的内部电阻的增加率。
进而制造了将添加硅烷偶联剂及作为碱性化合物的氢氧化锂的同时而制作的实施例9~实施例12的钛酸锂粒子群作为负极的活性物质层的锂离子二次电池。这些锂离子二次电池的制造方法及制造条件与使用实施例1~实施例8、比较例1~比较例5的钛酸锂粒子群的锂离子二次电池相同。然后,与使用未添加硅烷偶联剂的比较例1的钛酸锂粒子群的锂离子二次电池一起,测定了使用实施例9~实施例12的钛酸锂粒子群的锂离子二次电池的初始的内部电阻(dcir)以及2000次循环后的内部电阻的增加率。初始的内部电阻及内部电阻的增加率的测定方法与实施例1~实施例8及比较例1相同。
图11及12中示出所述测定结果。图11是表示硅烷偶联剂的添加量与初始的内部电阻(dcir)的关系的图表。图12是表示硅烷偶联剂的添加量与内部电阻的增加率的关系的图表。如图11所示,根据在添加硅烷偶联剂及碱性化合物的同时而制作的钛酸锂粒子群,与未添加碱性化合物的情况相比,确认了初始的内部电阻(dcir)的值没有变化或略有下降。另一方面,如图12所示,根据在添加硅烷偶联剂及碱性化合物的同时而制作的钛酸锂粒子群,与未添加碱性化合物的情况相比,确认了内部电阻的增加率得到进一步抑制。
例如,比较图12及图10可知,在硅烷偶联剂的添加量为3wt%的情况下,根据有无添加碱性化合物,内部电阻的增加率有21.6%的差。在硅烷偶联剂的添加量为5wt%的情况下,根据有无添加碱性化合物,内部电阻的增加率有7.5%的差。在硅烷偶联剂的添加量为10wt%的情况下,根据有无添加碱性化合物,内部电阻的增加率有10.4%的差。在硅烷偶联剂的添加量为20wt%的情况下,根据有无添加碱性化合物,内部电阻的增加率有6.9%的差。特别是,如果添加碱性化合物,硅烷偶联剂的添加量为20wt%时的内部电阻的增加率被抑制到与未添加碱性化合物且硅烷偶联剂的添加量为10wt%的情况相同。
即,确认了通过在制造工序中添加碱性化合物,提高了相对于制造过程中投入的硅烷偶联剂的量的内部电阻的增加率抑制效果。
继而,制作了将进行了使用氢氧化锂作为碱性化合物,与水及钛酸锂粒子混合后,过滤、干燥这样的前处理后,使用硅烷偶联剂制作的实施例13的钛酸锂粒子群作为负极的活性物质层的锂离子二次电池。这些锂离子二次电池的制造方法及制造条件与使用实施例1~实施例8、比较例1~比较例5的钛酸锂粒子群的锂离子二次电池相同。然后,测定了初始的内部电阻(dcir)以及2000次循环后的内部电阻的增加率。初始的内部电阻及内部电阻的增加率的测定方法与实施例1~实施例8及比较例1相同。
下表1中示出所述测定结果。下表1中同时记载了将实施例9的钛酸锂粒子群作为负极的活性物质层的锂离子二次电池的结果。实施例13中,在利用碱性化合物对钛酸锂粒子群进行前处理后转移到硅烷偶联剂的混合工序,与此相对,实施例9中,钛酸锂粒子群、碱性化合物及硅烷偶联剂混合,这一点上实施例9与实施例13不同。
(表1)
如表1所示,关于初始的内部电阻,在实施例9及实施例13中几乎未确认到差异。但是,在实施例13中,2000次循环后的内阻增加率更好。即,确认到如果预先利用碱性化合物对金属化合物粒子群进行前处理,则内部电阻的增加率抑制效果会进一步提高。
(关于比较例中的si量)
在比较例3~比较例6中,验证了内部电阻增加率的抑制效果低的理由。具体而言,在异丙醇中添加钛酸锂粒子群及硅烷偶联剂(10wt%)制成混合溶媒。将使用均化器搅拌混合溶媒而获得的凝聚物在150℃下干燥12小时。再者,未进行加热处理。使用异丙醇对干燥后的粉末清洗3次。将清洗后的粉末在150℃下干燥12小时。
图13(a)示出对干燥后的粉末及清洗后干燥的粉末进行热重量测定的结果。根据图13(a),清洗前的粉末由于加热而失去了5.24wt%的重量。另一方面,清洗后干燥的粉末因加热而失去的重量为1.51wt%。如果将所述1.51wt%的损失量换算成硅烷偶联剂的量,则如图13(b)所示,为1.7wt%。即,明确了清洗前含有10wt%的硅烷偶联剂在清洗后减少到1.7wt%。如此未进行加热处理的比较例3~比较例6中,可知硅烷偶联剂在溶媒中溶解,因此如上所述,内部电阻增加的抑制效果变低。
相对于钛酸锂粒子群总量的硅烷偶联剂的量为1.7wt%时,钛酸锂粒子群中含有的si元素的含量换算为0.3wt%。因此,根据图13所示的结果,通过添加10wt%的硅烷偶联剂,在不经过加热处理的情况下,可以说制作出以0.3wt%的比例含有si元素的钛酸锂粒子群。
(关于实施例中的si量)
测定了实施例1~实施例4以及比较例1的钛酸锂粒子群中残存的si元素的量。作为si元素的定量方法,使用了碱熔融法。即,在铂坩埚中加入实施例1~实施例4以及比较例1的钛酸锂粒子群与熔剂,通过加热使其熔融,进而添加酸,由此使实施例1~实施例4以及比较例1的钛酸锂粒子群溶解。然后,利用电感耦合等离子体原子发射光谱法(inductivelycoupledplasma-atomicemissionspectroscopy,icp-aes)(高频感应耦合等离子体发光能谱分析法)对si量进行定量。其结果如图14所示。图14是表示硅烷偶联剂的添加量与钛酸锂粒子群中含有的si元素的量的关系的图表。
如图14所示,在混合10wt%的硅烷偶联剂并在混合工序后进行加热处理的情况下,钛酸锂粒子群中残存的si元素的量为1.36wt%。在混合了相同量的硅烷偶联剂但在混合工序后未进行加热处理的比较例4中,钛酸锂粒子群中残存的si元素的量基于图9所示的结果为0.3wt%,因此可确认si含量飞跃性地提高。另外,确认了在添加3wt%以上的硅烷偶联剂的情况下,通过混合工序后的加热处理,残存有比比较例4多的si元素。
然后,合并图8所示的结果与图14所示的结果,如实施例1~实施例4所示,通过以3wt%以上且20wt%添加硅烷偶联剂,并在混合工序后进行加热处理,而可制作si元素的含量为0.4wt%以上且3.0wt%以下的钛酸锂粒子群,根据此钛酸锂粒子群,确认到内部电阻增加得到抑制。进而,合并图7所示的结果与图14所示的结果,如实施例1、实施例2及实施例4所示,通过以3wt%以上且10wt%添加硅烷偶联剂,并在混合工序后进行加热处理,而可制作si元素的含量为0.4wt%以上且1.4wt%以下的钛酸锂粒子群,根据此钛酸锂粒子群,确认到也可抑制初始的内部电阻。
(关于实施例中的si)
接着,在进行了加热处理的实施例1~实施例4中,对在钛酸锂粒子群的表面呈现出硅烷偶联剂的形态进行了验证。具体而言,为了确认加热处理中的硅烷偶联剂的反应,加热(ch3o)3sic3h6ococh=ch2的硅烷偶联剂而进行元素分析。具体而言,使在钛酸锂粒子群中混合了硅烷偶联剂(10wt%)而得的凝聚物在150℃下干燥12小时。干燥后,从300℃至500℃实施加热处理。使用碳分析仪对加热处理后的粉末进行碳重量测定。图15(a)是表示各加热温度下的硅烷偶联剂的碳浓度(wt%)的图表。图15(b)是表示各加热温度下的硅烷偶联剂的硅浓度(wt%)的图表。另外,图15(c)是表示各加热温度下的碳/硅比的图表。
由图15(a)明确可知,随着加热温度的上升,硅烷偶联剂的碳量下降。加热温度达到300℃时,碳量明显减少,在400℃时几乎消失。另一方面,由图15(b)明确可知,硅烷偶联剂的硅量与加热温度无关,基本呈固定的值。
参照基于这些结果制成的图15(c)的碳/硅比的图表也明确可知,推测通过加热工序,硅烷偶联剂的碳消失,成为了与硅氧化物极其接近的结构。即,可认为,硅烷偶联剂通过在300℃~600℃下加热,不是作为硅烷偶联剂,而是成为硅氧化物,在金属化合物粒子群的表面形成了涂布层。
接着,对混合了10wt%的硅烷偶联剂的钛酸锂粒子群进行了x射线光电能谱分析。图16是表示测定加热处理前的钛酸锂粒子群与加热处理后的钛酸锂粒子群的结果的图表。键合能103ev表示si-oh及si-o-c为构成官能基。另外,键合能104ev表示si-o-si及sio2是构成官能基。
由图16明确可知,加热处理前的钛酸锂粒子群的光谱在103ev与104ev的中间地点有峰值。另一方面,在加热处理后的钛酸锂粒子群中,峰值偏移至104ev附近。即,加热处理后的钛酸锂粒子群被认为也含有sio或si,但可知具有与sio2非常接近的峰值。
接着,图17示出对加热处理后的钛酸锂粒子群进行x射线衍射分析的结果。如图中虚线所示,sio2在24度附近有峰值。但是,在加热处理后的钛酸锂粒子群的光谱中,没有出现表示sio2的峰值。这是因为在x射线衍射分析中在24度附近检测出sio2的峰值的原因是结晶质的硅氧化物。即,虽然在x射线光电能谱分析中确认到了与sio2非常接近的峰值,但是在x射线衍射分析中没有确认到表示sio2的峰值,因此可知涂布层中含有的硅氧化物为非晶状。
(关于固体1h-nmr)
图18的(a)表示对所获得的实施例2及比较例1的钛酸锂粒子群进行测定的固体1h-nmr的数据。测定使用核磁共振分析装置(日本电子制造的jnm-eca600)进行。存在于5ppm~8ppm的峰值表示钛酸锂粒子群的表面羟基(桥羟基(bridgeoh))。
图13(b)中示出利用波形分离软件对图18(a)的5ppm~8ppm中存在的峰值计算积分强度的结果。由图18(b)也明确可知,与比较例1的钛酸锂粒子群的积分强度相比,实施例2的钛酸锂粒子群的积分强度减少了44%左右。即,表示存在于实施例2的钛酸锂粒子群的表面的羟基(桥羟基(bridgeoh))的峰值因硅化合物的附着而减少。换言之,可认为实施例2是硅氧化物被覆了未被覆硅氧化物的比较例1的钛酸锂粒子群的表面的44%左右的形态。
接着,图19表示使用实施例1、实施例2及比较例1的钛酸锂粒子群的锂离子二次电池在2000次循环后的内部电阻值的增加率。积分强度的减少率为29%的实施例1、及积分强度的减少率为44%的实施例2与比较例1的锂离子二次电池相比,均抑制了2000次循环后的内部电阻值的增加率,得到了良好的结果。认为这是由于钛酸锂粒子群表面的硅氧化物的被覆抑制了负极上的堆积物生成。就抑制内部电阻上升的观点而言,钛酸锂粒子群的积分强度的减少率理想为25%以上。
[符号的说明]
a:金属化合物粒子群
c:涂布层
1:一次粒子
2:空隙
- 还没有人留言评论。精彩留言会获得点赞!