一种3-氨基丙基三乙氧基硅烷改性氧化石墨烯/硝化棉复合物的制备方法与流程
本发明属于含能材料领域,具体涉及一种3-氨基丙基三乙氧基硅烷改性氧化石墨烯//硝化棉(nc)复合物的制备方法。
背景技术:
硝化棉(nc)作为传统的含能粘合剂,是双基推进剂的重要组分之一;在中小型火箭、导弹发动机装药中应用广泛。nc的热分解提供的能量远大于复合推进剂等推进剂中的粘结剂所提供的能量。同时,nc作为双基系固体推进剂的主要组分,其热分解对固体推进剂的能量水平有决定性的作用,提高nc的表观分解热,可以显著提高固体推进剂的能量水平。但由于nc分子链刚性较强等原因,nc的玻璃化温度较高,导致双基推进剂低温延伸率小,易脆化,限制了它的应用温度范围。
氧化石墨烯(go)是一种重要的石墨烯衍生物,在复合材料领域有着独特的用途。其表面上存在大量的含氧基团,如羟基、羧基、环氧基等,可以通过共价或非共价的方法在氧化石墨烯表面进行功能化修饰,得到功能化氧化石墨烯,功能化后的改性氧化石墨烯可与橡胶、聚乙烯醇、壳聚糖等高分子形成作用力,用于改性高分子材料的特性,也可与纳米金属氧化物作用,用于太阳能电池。xinzhang等在appliedphysicsletters,2013,102:141905~141909《directlaserinitiationandimprovedthermalstabilityofnitrocellulose/grapheneoxidenanocomposites》利用溶剂-非溶剂法公开制备了go/nc复合材料,研究了go对nc热分解过程的影响。此方法是主要研究了go/nc复合材料的燃烧速度,以及活化能对热分解温度的影响,并未给出go对nc其它性能如玻璃化温度的相关技术启示。。袁申等在含能材料,2017,25(3):203~208《ngo/nc复合含能材料的制备及热分解性能》一文公开制备了硝基石墨烯(ngo)/nc复合含能材料,研究了ngo对nc热分解的催化性能。其中,ngo/nc复合物较nc而言,放热峰温度由201℃提高至213℃。但此方法主要是改善nc的热性能,提高了nc的放热峰温度,也未直接给出该方法对nc玻璃化温度影响的相关技术启示。
技术实现要素:
本发明要解决的技术问题是克服背景技术中的不足和缺陷,提供一种既改善了nc热性能,也降低了nc玻璃化温度的3-氨基丙基三乙氧基硅烷改性氧化石墨烯/硝化棉(nc)复合物的制备方法。
为了实现上述技术任务,本发明采用如下技术方案予以实现:
一种3-氨基丙基三乙氧基硅烷改性氧化石墨烯/硝化棉(nc)复合物的制备方法,其包括如下步骤:
步骤2,将步骤1制备的3-氨基丙基三乙氧基硅烷改性氧化石墨烯加入四氢呋喃,在20~35℃的温度范围内超声分散1~2h,其中3-氨基丙基三乙氧基硅烷改性氧化石墨烯与四氢呋喃用量为10mg~60mg:20g~150g,得到3-氨基丙基三乙氧基硅烷改性氧化石墨烯分散液;
步骤3,将nc加入到四氢呋喃中,在20~35℃的温度范围内搅拌1~2h,其中nc和四氢呋喃的用量比4.0g~6.0g:100g~200g,待nc完全溶解,得到nc与四氢呋喃混合溶液;
步骤4,将步骤2得到的3-氨基丙基三乙氧基硅烷改性氧化石墨烯分散液倒入nc混合溶液中,20~40℃下对体系搅拌0.5~1h,待均匀,常温放置1~2个星期,在30~40℃下烘干2~4h得到3-氨基丙基三乙氧基硅烷改性氧化石墨烯/硝化棉(nc)复合物。
进一步地,本发明的步骤1,制备3-氨基丙基三乙氧基硅烷改性氧化石墨烯,具体包括如下步骤:
步骤1-1,将氧化石墨烯与四氢呋喃按照20~100mg:35.6g~222.5g进行混合;
步骤1-2,将步骤1-1得到的混合物,在20~35℃的温度范围内超声分散1~2h后,再加入3-氨基丙基三乙氧基硅烷,其中氧化石墨烯、四氢呋喃按照、3-氨基丙基三乙氧基硅烷的质量比为20~100mg:35.6g~222.5g:3.76*10-3mg~2.82*10-2mg;
步骤1-3,将步骤1-2得到的混合反应物,在60~70℃的温度范围内搅拌反应6~10h,离心,洗涤,干燥,得到粉末状3-氨基丙基三乙氧基硅烷改性氧化石墨烯。
本发明与现有技术相比,有益技术效果如下:
本发明的3-氨基丙基三乙氧基硅烷改性氧化石墨烯//nc复合物不仅可以改善nc的热性能,热分解温度提高0.5℃~6.6℃,还可以降低nc的玻璃化温度,降低10.5℃~19.8℃。本发明能够有效降低硝化棉的玻璃化温度,可以拓宽其温度适应范围,有利于其在低温环境中的应用。
附图说明
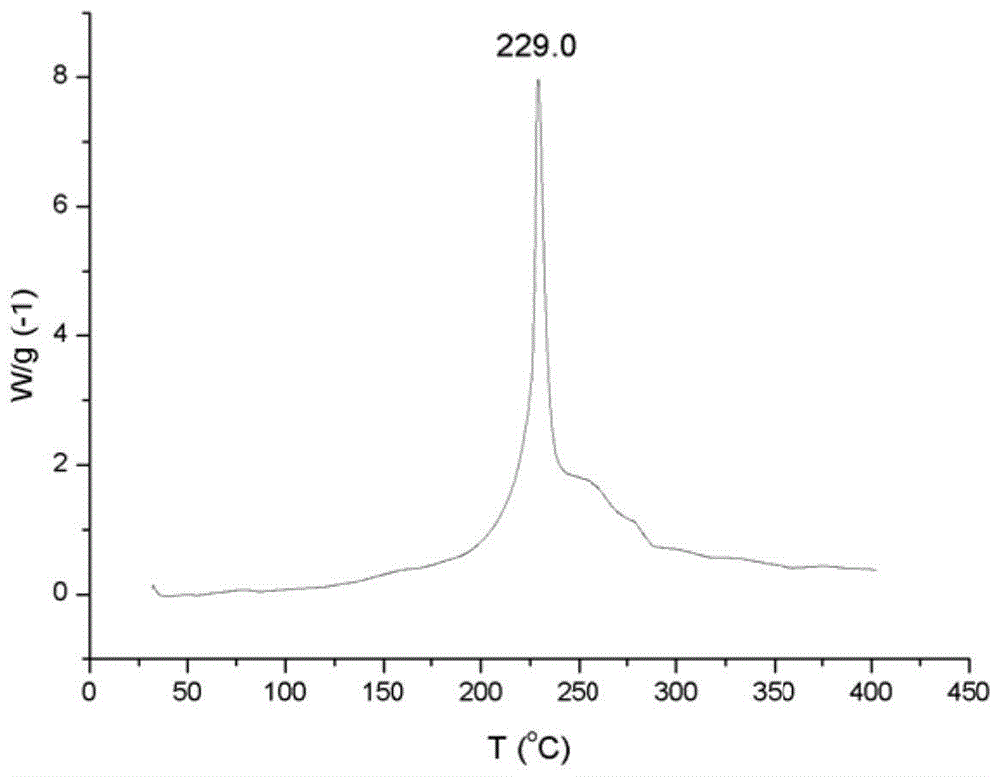
图1是本发明涉及的实施例制备原料nc热分解图。
图2是本发明涉及的实施例制备的原料nc玻璃化温度图。
图3是本发明的实施例1制备的氧化石墨烯/nc复合物热分解图。
图4是本发明的实施例1制备的氧化石墨烯/nc复合物玻璃化温度图。
图5是本发明涉及的实施例制备原料nc的sem图。
图6是本发明的实施例1制备的氧化石墨烯/nc复合物sem图。
图7是本发明涉及的实施例制备原料nc红外图。
图8是本发明的实施例1制备的氧化石墨烯/nc复合物红外图。
图9是本发明实施例15制备的3-氨基丙基三乙氧基硅烷改性氧化石墨烯红外图。
图10是本发明实施例15制备的3-氨基丙基三乙氧基硅烷改性氧化石墨烯xps图。
图11是本发明实施例15制备的3-氨基丙基三乙氧基硅烷改性氧化石墨烯sem图。
图12是本发明的实施例15制备的3-氨基丙基三乙氧基硅烷改性氧化石墨烯拉曼图。
图13是本发明实施例15还原的3-氨基丙基三乙氧基硅烷改性石墨烯sem图。
以下结合附图及具体实施方式对本发明的具体内容作进一步详细说明。
具体实施方式
本发明所用的氧化石墨烯通过经销商北京百灵威科技有限公司购买。硝化棉(nc),由西安近代化学研究所市售产品,其中氮含量11.2%~12%,热分解温度229.0℃,玻璃化温度-64.1℃。其中图1和2分别为制备的原料nc的热分解图和玻璃化温度图。
第一部分关于3-氨基丙基三乙氧基硅烷改性氧化石墨烯/硝化棉复合物
实施例1
将20mg3-氨基丙基三乙氧基硅烷改性氧化石墨烯加入60g四氢呋喃中,在25℃超声分散1.5h,将nc4.0g加入到100g四氢呋喃中,在23℃搅拌1.5h,待nc完全溶解;将氨基功能化氧化石墨烯分散液倒入nc溶液中,30℃下对体系搅拌1h,待均匀后倒入模具中。常温放置2个星期后,在35℃下烘干3.0h得到相应的氨基功能化氧化石墨烯/硝化棉(nc)复合物4.0g。氨基功能化氧化石墨烯/nc复合物热分解温度为235.6℃,较nc提高了6.6℃,玻璃化温度为-83.9℃,较nc降低了19.8℃。其中图3和4分别为实施例1制备的氨基功能化氧化石墨烯/nc复合物热分解图和玻璃化温度图。
结构分析
1.扫描电镜(sem)分析
发现原来的nc直接有大量的空隙,加入了石墨烯和功能化石墨烯后,空隙变少或者没有,并且可以看见硝化棉包裹球状物在表面,应该是硝化棉包裹的氧化石墨烯。图5是本发明涉及的实施例制备原料nc的sem图。图6是实施例1制备的氨基功能化氧化石墨烯/nc复合物的sem图。
2.红外分析
氨基功能化氧化石墨烯/nc复合物的红外图谱与/nc复合物的红外图谱相似,因为氨基功能化氧化石墨烯加入量少,其特征峰如si-o键在红外中不明显,而其它的特征峰如羟基、羰基、烷氧基等官能团,nc中也有。图9是本发明涉及的实施例制备原料nc的红外图。图10是实施例1制备的氨基功能化氧化石墨烯/nc复合物的红外图。
实施例2
将60mg3-氨基丙基三乙氧基硅烷改性氧化石墨烯加入150g四氢呋喃中,在35℃超声分散2.0h,将nc6.0g加入到200g四氢呋喃中,在34℃搅拌2.0h,待nc完全溶解;将氨基功能化氧化石墨烯分散液倒入nc溶液中,39℃下对体系搅拌1h,待均匀后倒入模具中。常温放置2个星期后,在38℃下烘干4.0h得到相应的氨基功能化氧化石墨烯/硝化棉(nc)复合物6.0g。氨基功能化氧化石墨烯/nc复合物热分解温度为230.1℃,玻璃化温度为-75.9℃。
实施例3
将55mg3-氨基丙基三乙氧基硅烷改性氧化石墨烯加入125g四氢呋喃中,在33℃超声分散1.0h,将nc5.9g加入到190g四氢呋喃中,在33℃搅拌1.5h,待nc完全溶解;将氨基功能化氧化石墨烯分散液倒入nc溶液中,36℃下对体系搅拌0.8h,待均匀后倒入模具中。常温放置1个星期后,在37℃下烘3.5h得到相应的氨基功能化氧化石墨烯/硝化棉(nc)复合物5.9g。氨基功能化氧化石墨烯/nc复合物热分解温度为235.2℃,玻璃化温度为-80.4℃。
实施例4
将51mg3-氨基丙基三乙氧基硅烷改性氧化石墨烯加入118g四氢呋喃中,在24℃超声分散1.5h,将nc4.9g加入到195g四氢呋喃中,在31℃搅拌1.8h,待nc完全溶解;将氨基功能化氧化石墨烯分散液倒入nc溶液中,32℃下对体系搅拌0.6h,待均匀后倒入模具中。常温放置1.5个星期后,在36℃下烘3.1h得到相应的氨基功能化氧化石墨烯/硝化棉(nc)复合物4.9g。氨基功能化氧化石墨烯/nc复合物热分解温度为231.9℃,玻璃化温度为-76.9℃。
实施例5
将47mg3-氨基丙基三乙氧基硅烷改性氧化石墨烯加入124g四氢呋喃中,在28℃超声分散1.6h,将nc5.5g加入到170g四氢呋喃中,在30℃搅拌1.5h,待nc完全溶解;将氨基功能化氧化石墨烯分散液倒入nc溶液中,29℃下对体系搅拌0.5h,待均匀后倒入模具中。常温放置2个星期后,在35℃下烘3.5h得到相应的氨基功能化氧化石墨烯/硝化棉(nc)复合物5.5g。氨基功能化氧化石墨烯/nc复合物热分解温度为234.4℃,玻璃化温度为-80.7℃。
实施例6
将41mg3-氨基丙基三乙氧基硅烷改性氧化石墨烯加入107g四氢呋喃中,在25℃超声分散2h,将nc3.9g加入到160g四氢呋喃中,在26℃搅拌1.3h,待nc完全溶解;将氨基功能化氧化石墨烯分散液倒入nc溶液中,33℃下对体系搅拌0.8h,待均匀后倒入模具中。常温放置1.6个星期后,在30℃下烘3h得到相应的氨基功能化氧化石墨烯/硝化棉(nc)复合物3.9g。氨基功能化氧化石墨烯/nc复合物热分解温度为231.8℃,玻璃化温度为-77.4℃。
实施例7
将36mg3-氨基丙基三乙氧基硅烷改性氧化石墨烯加入88g四氢呋喃中,在27℃超声分散1.5h,将nc5.4g加入到190g四氢呋喃中,在29℃搅拌1.5h,待nc完全溶解;将氨基功能化氧化石墨烯分散液倒入nc溶液中,38℃下对体系搅拌0.9h,待均匀后倒入模具中。常温放置1.4个星期后,在30℃下烘2.5h得到相应的氨基功能化氧化石墨烯/硝化棉(nc)复合物5.4g。氨基功能化氧化石墨烯/nc复合物热分解温度为230.2℃,玻璃化温度为-77.3℃。
实施例8
将30mg3-氨基丙基三乙氧基硅烷改性氧化石墨烯加入96g四氢呋喃中,在31℃超声分散1.8h,将nc6.0g加入到195g四氢呋喃中,在20℃搅拌1.4h,待nc完全溶解;将氨基功能化氧化石墨烯分散液倒入nc溶液中,35℃下对体系搅拌1.0h,待均匀后倒入模具中。常温放置2个星期后,在36℃下烘3h得到相应的氨基功能化氧化石墨烯/硝化棉(nc)复合物6.0g。氨基功能化氧化石墨烯/nc复合物热分解温度为232.4℃,玻璃化温度为-81.2℃。
实施例9
将27mg3-氨基丙基三乙氧基硅烷改性氧化石墨烯加入67g四氢呋喃中,在22℃超声分散1.0h,将nc4.2g加入到104g四氢呋喃中,在26℃搅拌1.4h,待nc完全溶解;将氨基功能化氧化石墨烯分散液倒入nc溶液中,32℃下对体系搅拌0.6h,待均匀后倒入模具中。常温放置1个星期后,在35℃下烘2.5h得到相应的氨基功能化氧化石墨烯/硝化棉(nc)复合物4.2g。氨基功能化氧化石墨烯/nc复合物热分解温度为222.7℃,玻璃化温度为-82.8℃。
实施例10
将24mg3-氨基丙基三乙氧基硅烷改性氧化石墨烯加入58g四氢呋喃中,在26℃超声分散1.8h,将nc5.7g加入到187g四氢呋喃中,在24℃搅拌2.0h,待nc完全溶解;将氨基功能化氧化石墨烯分散液倒入nc溶液中,35℃下对体系搅拌0.5h,待均匀后倒入模具中。常温放置1.6个星期后,在32℃下烘3.4h得到相应的氨基功能化氧化石墨烯/硝化棉(nc)复合物5.7g。氨基功能化氧化石墨烯/nc复合物热分解温度为230.2℃,玻璃化温度为-74.9℃。
实施例11
将20mg3-氨基丙基三乙氧基硅烷改性氧化石墨烯加入46g四氢呋喃中,在29℃超声分散1.4h,将nc4.1g加入到115g四氢呋喃中,在26℃搅拌1.4h,待nc完全溶解;将氨基功能化氧化石墨烯分散液倒入nc溶液中,27℃下对体系搅拌0.7h,待均匀后倒入模具中。常温放置1.2个星期后,在31℃下烘2.4h得到相应的氨基功能化氧化石墨烯/硝化棉(nc)复合物4.1g。氨基功能化氧化石墨烯/nc复合物热分解温度为229.5℃,玻璃化温度为-74.6℃。
实施例12
将16mg3-氨基丙基三乙氧基硅烷改性氧化石墨烯加入32g四氢呋喃中,在26℃超声分散1.6h,将nc5.2g加入到150g四氢呋喃中,在35℃搅拌1.4h,待nc完全溶解;将氨基功能化氧化石墨烯分散液倒入nc溶液中,34℃下对体系搅拌0.8h,待均匀后倒入模具中。常温放置2个星期后,在33℃下烘3.5h得到相应的氨基功能化氧化石墨烯/硝化棉(nc)复合物5.2g。氨基功能化氧化石墨烯/nc复合物热分解温度为234.1℃,玻璃化温度为-77.8℃。
实施例13
将13mg3-氨基丙基三乙氧基硅烷改性氧化石墨烯加入28g四氢呋喃中,在20℃超声分散2.0h,将nc4.0g加入到114g四氢呋喃中,在21℃搅拌1.1h,待nc完全溶解;将氨基功能化氧化石墨烯分散液倒入nc溶液中,27℃下对体系搅拌0.6h,待均匀后倒入模具中。常温放置1个星期后,在32℃下烘2.1h得到相应的氨基功能化氧化石墨烯/硝化棉(nc)复合物4.0g。氨基功能化氧化石墨烯/nc复合物热分解温度为230.8℃,玻璃化温度为-78.2℃。
实施例14
将10mg3-氨基丙基三乙氧基硅烷改性氧化石墨烯加入20g四氢呋喃中,在23℃超声分散1.5h,将nc5.0g加入到124g四氢呋喃中,在30℃搅拌1.5h,待nc完全溶解;将氨基功能化氧化石墨烯分散液倒入nc溶液中,30℃下对体系搅拌0.5h,待均匀后倒入模具中。常温放置1.7个星期后,在36℃下烘3.6h得到相应的氨基功能化氧化石墨烯/硝化棉(nc)复合物5.0g。氨基功能化氧化石墨烯/nc复合物热分解温度为235.4℃,玻璃化温度为-76.8℃。
第二部分关于3-氨基丙基三乙氧基硅烷改性氧化石墨烯制备
本发明的3-氨基丙基三乙氧基硅烷改性氧化石墨烯/硝化棉复合物的制备方法,其中步骤1制备3-氨基丙基三乙氧基硅烷改性氧化石墨烯,还包括如下步骤:
步骤1-1,将氧化石墨烯与四氢呋喃按照质量比20~100mg:35.6g~222.5g进行混合;
步骤1-2,将步骤1-1得到的混合物,在20~35℃的温度范围内超声分散1~2h后,再加入3-氨基丙基三乙氧基硅烷,其中氧化石墨烯、四氢呋喃按照、3-氨基丙基三乙氧基硅烷的质量比为20~100mg:35.6g~222.5g:3.76*10-3mg~2.82*10-2mg;
步骤1-3,将步骤1-2得到的混合反应物,在60~70℃的温度范围内搅拌反应6~8h,离心,洗涤,干燥,得到粉末状3-氨基丙基三乙氧基硅烷改性氧化石墨烯。
为了更好的验证本发明给出的3-氨基丙基三乙氧基硅烷改性氧化石墨烯制备方法的可靠性,申请人也进行了大量的实验进行验证,最终证明该方法涉及的配方可行性,以及对最终产物3-氨基丙基三乙氧基硅烷改性氧化石墨烯/硝化棉复合物的效果一致性。
申请人也为此给出一系列制备3-氨基丙基三乙氧基硅烷改性氧化石墨烯的制备实施例,具体如下:
实施例15
3-氨基丙基三乙氧基硅烷改性氧化石墨烯的合成
将20mg氧化石墨烯加入35.6g四氢呋喃中,在25℃超声分散1.5h,然后加入3.76*10-3mg3-氨基丙基三乙氧基硅烷,70℃反应8.0h,离心,洗涤,干燥,得到黑色粉末3-氨基丙基三乙氧基硅烷改性氧化石墨烯30mg。
结构鉴定:
1.红外分析
在目标化合物3-氨基丙基三乙氧基硅烷改性氧化石墨烯的红外光谱中可以看出,氧化石墨中1740cm-1处的羰基伸缩振动吸收峰已经移至1636cm-1,相应的氧化石墨中1248cm-1处的环氧基特征吸收峰变得非常弱,甚至消失,说明3-氨基丙基三乙氧基硅烷中的部分氨基与氧化石墨中的环氧基发生了加成反应。改性氧化石墨在1040cm-1处出现了si-o-si键的伸缩振动吸收峰,这是由3-氨基丙基三乙氧基硅烷中的部分烷氧基经水解缩合而形成的。说明氧化石墨烯表面已被3-氨基丙基三乙氧基硅烷改性。图9是实施例15制备的3-巯基丙基三乙氧基硅烷改性氧化石墨烯红外图。
2.x射线光电子能谱(xps)分析
xps谱图显示,氧化石墨烯只含有289ev和535ev两个c1s和o1s特征峰。对比氧化石墨烯和发现3-氨基丙基三乙氧基硅烷改性氧化石墨烯的表征结果,发现3-氨基丙基三乙氧基硅烷改性氧化石墨烯除了c1s和o1s特征峰外,还在402ev和102ev处出现新的n1s和si2p谱峰。该结果证明3-氨基丙基三乙氧基硅烷成功接枝在氧化石墨烯结构中。图10是实施例15制备的3-氨基丙基三乙氧基硅烷改性氧化石墨烯xps图。
3.扫描电镜(sem)分析
分析电镜结果,3-氨基丙基三乙氧基硅烷改性氧化石墨烯的片状结构明显存在,未发生大规模团聚。并且功能化后,3-氨基丙基三乙氧基硅烷改性氧化石墨烯片层上面的褶皱明显变多。图11是实施例15制备的3-氨基丙基三乙氧基硅烷改性氧化石墨烯sem图。
4.拉曼分析
nh-si-fgo的拉曼图谱显示其d峰和g峰分别出现在1355cm-1和1599cm-1。拉曼光谱的d带与g带的强度比也表示sp3/sp2碳原子比。其中,nh-si-fgo的id/ig=1.119,高于go的id/ig=1.027。这是由于go被功能化后,sp3杂环碳原子增多的原因。图12是实施例15制备的3-氨基丙基三乙氧基硅烷改性氧化石墨烯拉曼图。
3-氨基丙基三乙氧基硅烷改性氧化石墨烯的还原
将洗涤后烘干的20mg3-氨基丙基三乙氧基硅烷改性氧化石墨烯分散于40ml四氢呋喃中,超声0.5h后,加入0.5g水合肼,于70℃下还原6h;再用无水乙醇和蒸馏水洗涤所得产物,并烘干,得到3-氨基丙基三乙氧基硅烷改性石墨烯15mg。称取10mg烘干后的3-氨基丙基三乙氧基硅烷改性石墨烯,分别分散于dmf,dmso,乙醇,thf,丙酮中,超声0.5~2h后,得到稳定的分散液,24h后不会出现沉淀和分层。图13是实施例15还原的3-氨基丙基三乙氧基硅烷改性石墨烯sem图。
实施例16
3-氨基丙基三乙氧基硅烷改性氧化石墨烯的合成
将40mg氧化石墨烯加入100g四氢呋喃中,在25℃超声分散1h,然后加入9*10-3mg3-氨基丙基三乙氧基硅烷,在60℃反应7.0h,离心,洗涤,干燥,得到黑色粉末3-氨基丙基三乙氧基硅烷改性氧化石墨烯58mg。
3-氨基丙基三乙氧基硅烷改性氧化石墨烯的还原
将洗涤后烘干的20mg3-氨基丙基三乙氧基硅烷改性氧化石墨烯分散于40ml四氢呋喃中,超声0.5h后,加入0.5g水合肼,于70℃下还原6h;再用无水乙醇和蒸馏水洗涤所得产物,并烘干,得到3-氨基丙基三乙氧基硅烷改性石墨烯。称取10mg烘干后的3-氨基丙基三乙氧基硅烷改性石墨烯,分别分散于dmf,dmso,乙醇,thf,丙酮中,超声0.5~2h后,得到稳定的分散液,24h后不会出现沉淀和分层。从该实施例的3-氨基丙基三乙氧基硅烷改性石墨烯的sem图结果中同样发现,还原后的石墨烯未出现团聚现象。
实施例17
3-氨基丙基三乙氧基硅烷改性氧化石墨烯的合成
将60mg氧化石墨烯加入140g四氢呋喃中,在30℃下超声分散2h,然后加入1.00*10-2mg3-氨基丙基三乙氧基硅烷,在65℃反应7h,离心,洗涤,干燥,得到黑色粉末3-氨基丙基三乙氧基硅烷改性氧化石墨烯85mg。
3-氨基丙基三乙氧基硅烷改性氧化石墨烯的还原
将洗涤后烘干的20mg3-氨基丙基三乙氧基硅烷改性氧化石墨烯分散于40ml四氢呋喃中,超声0.5h后,加入0.5g水合肼,于70℃下还原6h;再用无水乙醇和蒸馏水洗涤所得产物,并烘干,得到3-氨基丙基三乙氧基硅烷改性石墨烯。称取10mg烘干后的3-氨基丙基三乙氧基硅烷改性石墨烯,分别分散于dmf,dmso,乙醇,thf,丙酮中,超声0.5~2h后,得到稳定的分散液,24h后不会出现沉淀和分层。从该实施例的3-氨基丙基三乙氧基硅烷改性石墨烯的sem图sem图结果中同样看出,还原后的石墨烯未出现团聚现象。
实施例18
3-氨基丙基三乙氧基硅烷改性氧化石墨烯的合成
将80mg氧化石墨烯加入190g四氢呋喃中,在35℃的温度下超声分散1.0h,然后加入1.63*10-2mg3-氨基丙基三乙氧基硅烷,在68℃反应7.5h,离心,洗涤,干燥,得到黑色粉末3-氨基丙基三乙氧基硅烷改性氧化石墨烯114mg。
3-氨基丙基三乙氧基硅烷改性氧化石墨烯的还原
将洗涤后烘干的20mg3-氨基丙基三乙氧基硅烷改性氧化石墨烯分散于40ml四氢呋喃中,超声0.5h后,加入0.5g水合肼,于70℃下还原6h;再用无水乙醇和蒸馏水洗涤所得产物,并烘干,得到3-氨基丙基三乙氧基硅烷改性石墨烯。称取10mg烘干后的3-氨基丙基三乙氧基硅烷改性石墨烯,分别分散于dmf,dmso,乙醇,thf,丙酮中,超声0.5~2h后,得到稳定的分散液,24h后不会出现沉淀和分层。从该实施例的3-氨基丙基三乙氧基硅烷改性石墨烯的sem图sem图结果中同样看出,还原后的石墨烯未出现团聚现象。
实施例19
3-氨基丙基三乙氧基硅烷改性氧化石墨烯的合成
将100mg氧化石墨烯加入222.5g四氢呋喃中,在30℃超声分散1.0h,然后加入2.82*10-2mg3-氨基丙基三乙氧基硅烷,在69℃反应7.0h,离心,洗涤,干燥,得到黑色粉末3-氨基丙基三乙氧基硅烷改性氧化石墨烯149mg。
3-氨基丙基三乙氧基硅烷改性氧化石墨烯的还原
将洗涤后烘干的20mg3-氨基丙基三乙氧基硅烷改性氧化石墨烯分散于40ml四氢呋喃中,超声0.5h后,加入0.5g水合肼,于70℃下还原6h;再用无水乙醇和蒸馏水洗涤所得产物,并烘干,得到3-氨基丙基三乙氧基硅烷改性石墨烯。称取10mg烘干后的3-氨基丙基三乙氧基硅烷改性石墨烯,分别分散于dmf,dmso,乙醇,thf,丙酮中,超声0.5~2h后,得到稳定的分散液,24h后不会出现沉淀和分层。从该实施例的3-氨基丙基三乙氧基硅烷改性石墨烯的sem图结果中同样看出,还原后的石墨烯未出现团聚现象。
实施例20
3-氨基丙基三乙氧基硅烷改性氧化石墨烯的合成
将90mg氧化石墨烯加入200g四氢呋喃中,在32℃超声分散1.2h,然后加入2.69*10-2mg3-氨基丙基三乙氧基硅烷,在67℃反应7.5h,离心,洗涤,干燥,得到黑色粉末3-氨基丙基三乙氧基硅烷改性氧化石墨烯130mg。
3-氨基丙基三乙氧基硅烷改性氧化石墨烯的还原
将洗涤后烘干的20mg3-氨基丙基三乙氧基硅烷改性氧化石墨烯分散于40ml四氢呋喃中,超声0.5h后,加入0.5g水合肼,于70℃下还原6h;再用无水乙醇和蒸馏水洗涤所得产物,并烘干,得到3-氨基丙基三乙氧基硅烷改性石墨烯。称取10mg烘干后的3-氨基丙基三乙氧基硅烷改性石墨烯,分别分散于dmf,dmso,乙醇,thf,丙酮中,超声0.5~2h后,得到稳定的分散液,24h后不会出现沉淀和分层。从该实施例的3-氨基丙基三乙氧基硅烷改性石墨烯的sem图中同样可以看出,还原后的石墨烯未出现团聚现象。
实施例21
3-氨基丙基三乙氧基硅烷改性氧化石墨烯的合成
将85mg氧化石墨烯加入184g四氢呋喃中,在33℃超声分散1.6h,然后加入2.47*10-2mg3-氨基丙基三乙氧基硅烷,在68℃反应7.2h,离心,洗涤,干燥,得到黑色粉末3-氨基丙基三乙氧基硅烷改性氧化石墨烯124mg。
3-氨基丙基三乙氧基硅烷改性氧化石墨烯的还原
将洗涤后烘干的20mg3-氨基丙基三乙氧基硅烷改性氧化石墨烯分散于40ml四氢呋喃中,超声0.5h后,加入0.5g水合肼,于70℃下还原6h;再用无水乙醇和蒸馏水洗涤所得产物,并烘干,得到3-氨基丙基三乙氧基硅烷改性石墨烯。称取10mg烘干后的3-氨基丙基三乙氧基硅烷改性石墨烯,分别分散于dmf,dmso,乙醇,thf,丙酮中,超声0.5~2h后,得到稳定的分散液,24h后不会出现沉淀和分层。从该实施例的3-氨基丙基三乙氧基硅烷改性石墨烯的sem图中同样可以看出,还原后的石墨烯未出现团聚现象。
实施例22
3-氨基丙基三乙氧基硅烷改性氧化石墨烯的合成
将95mg氧化石墨烯加入217g四氢呋喃中,在22℃超声分散1.4h,然后加入2.64*10-2mg3-氨基丙基三乙氧基硅烷,在66℃反应7.3h,离心,洗涤,干燥,得到黑色粉末3-氨基丙基三乙氧基硅烷改性氧化石墨烯140mg。
3-氨基丙基三乙氧基硅烷改性氧化石墨烯的还原
将洗涤后烘干的20mg3-氨基丙基三乙氧基硅烷改性氧化石墨烯分散于40ml四氢呋喃中,超声0.5h后,加入0.5g水合肼,于70℃下还原6h;再用无水乙醇和蒸馏水洗涤所得产物,并烘干,得到3-氨基丙基三乙氧基硅烷改性石墨烯。称取10mg烘干后的3-氨基丙基三乙氧基硅烷改性石墨烯,分别分散于dmf,dmso,乙醇,thf,丙酮中,超声0.5~2h后,得到稳定的分散液,24h后不会出现沉淀和分层。从该实施例的3-氨基丙基三乙氧基硅烷改性石墨烯的sem图中可以看出,还原后的石墨烯未出现团聚现象。
实施例23
3-氨基丙基三乙氧基硅烷改性氧化石墨烯的合成
将80mg氧化石墨烯加入174.9g四氢呋喃中,在21℃超声分散1.3h,然后加入2.01*10-2mg3-氨基丙基三乙氧基硅烷,在64℃反应6.4h,离心,洗涤,干燥,得到黑色粉末3-氨基丙基三乙氧基硅烷改性氧化石墨烯114mg。
3-氨基丙基三乙氧基硅烷改性氧化石墨烯的还原
将洗涤后烘干的20mg3-氨基丙基三乙氧基硅烷改性氧化石墨烯分散于40ml四氢呋喃中,超声0.5h后,加入0.5g水合肼,于70℃下还原6h;再用无水乙醇和蒸馏水洗涤所得产物,并烘干,得到3-氨基丙基三乙氧基硅烷改性石墨烯。称取10mg烘干后的3-氨基丙基三乙氧基硅烷改性石墨烯,分别分散于dmf,dmso,乙醇,thf,丙酮中,超声0.5~2h后,得到稳定的分散液,24h后不会出现沉淀和分层。从该实施例的3-氨基丙基三乙氧基硅烷改性石墨烯的sem图中同样可以看出,还原后的石墨烯未出现团聚现象。
实施例24
3-氨基丙基三乙氧基硅烷改性氧化石墨烯的合成
将77mg氧化石墨烯加入180g四氢呋喃中,在24℃超声分散1.0h,然后加入1.99*10-2mg3-氨基丙基三乙氧基硅烷,在64℃反应7.0h,离心,洗涤,干燥,得到黑色粉末3-氨基丙基三乙氧基硅烷改性氧化石墨烯106mg。
3-氨基丙基三乙氧基硅烷改性氧化石墨烯的还原
将洗涤后烘干的20mg3-氨基丙基三乙氧基硅烷改性氧化石墨烯分散于40ml四氢呋喃中,超声0.5h后,加入0.5g水合肼,于70℃下还原6h;再用无水乙醇和蒸馏水洗涤所得产物,并烘干,得到3-氨基丙基三乙氧基硅烷改性石墨烯。称取10mg烘干后的3-氨基丙基三乙氧基硅烷改性石墨烯,分别分散于dmf,dmso,乙醇,thf,丙酮中,超声0.5~2h后,得到稳定的分散液,24h后不会出现沉淀和分层。从该实施例的3-氨基丙基三乙氧基硅烷改性石墨烯的sem图中同样可以看出,还原后的石墨烯未出现团聚现象。
实施例25
3-氨基丙基三乙氧基硅烷改性氧化石墨烯的合成
将73mg氧化石墨烯加入177g四氢呋喃中,在26℃超声分散1.1h,然后加入1.80*10-2mg3-氨基丙基三乙氧基硅烷,在62℃反应6.5h,离心,洗涤,干燥,得到黑色粉末3-氨基丙基三乙氧基硅烷改性氧化石墨烯105mg。
3-氨基丙基三乙氧基硅烷改性氧化石墨烯的还原
将洗涤后烘干的20mg3-氨基丙基三乙氧基硅烷改性氧化石墨烯分散于40ml四氢呋喃中,超声0.5h后,加入0.5g水合肼,于70℃下还原6h;再用无水乙醇和蒸馏水洗涤所得产物,并烘干,得到3-氨基丙基三乙氧基硅烷改性石墨烯。称取10mg烘干后的3-氨基丙基三乙氧基硅烷改性石墨烯,分别分散于dmf,dmso,乙醇,thf,丙酮中,超声0.5~2h后,得到稳定的分散液,24h后不会出现沉淀和分层。从该实施例的3-氨基丙基三乙氧基硅烷改性石墨烯的sem图中同样可以看出,还原后的石墨烯未出现团聚现象。
实施例26
3-氨基丙基三乙氧基硅烷改性氧化石墨烯的合成
将69mg氧化石墨烯加入174g四氢呋喃中,在27℃超声分散1.2h,然后加入1.74*10-2mg3-氨基丙基三乙氧基硅烷,在61℃反应6.3h,离心,洗涤,干燥,得到黑色粉末3-氨基丙基三乙氧基硅烷改性氧化石墨烯103mg。
3-氨基丙基三乙氧基硅烷改性氧化石墨烯的还原
将洗涤后烘干的20mg3-氨基丙基三乙氧基硅烷改性氧化石墨烯分散于40ml四氢呋喃中,超声0.5h后,加入0.5g水合肼,于70℃下还原6h;再用无水乙醇和蒸馏水洗涤所得产物,并烘干,得到3-氨基丙基三乙氧基硅烷改性石墨烯。称取10mg烘干后的3-氨基丙基三乙氧基硅烷改性石墨烯,分别分散于dmf,dmso,乙醇,thf,丙酮中,超声0.5~2h后,得到稳定的分散液,24h后不会出现沉淀和分层。从该实施例的3-氨基丙基三乙氧基硅烷改性石墨烯的sem图中同样可以看出,还原后的石墨烯未出现团聚现象。
实施例27
3-氨基丙基三乙氧基硅烷改性氧化石墨烯的合成
将64mg氧化石墨烯加入169g四氢呋喃中,在34℃超声分散1.6h,然后加入1.71*10-2mg3-氨基丙基三乙氧基硅烷,在64℃反应7.6h,离心,洗涤,干燥,得到黑色粉末3-氨基丙基三乙氧基硅烷改性氧化石墨烯93mg。
3-氨基丙基三乙氧基硅烷改性氧化石墨烯的还原
将洗涤后烘干的20mg3-氨基丙基三乙氧基硅烷改性氧化石墨烯分散于40ml四氢呋喃中,超声0.5h后,加入0.5g水合肼,于70℃下还原6h;再用无水乙醇和蒸馏水洗涤所得产物,并烘干,得到3-氨基丙基三乙氧基硅烷改性石墨烯。称取10mg烘干后的3-氨基丙基三乙氧基硅烷改性石墨烯,分别分散于dmf,dmso,乙醇,thf,丙酮中,超声0.5~2h后,得到稳定的分散液,24h后不会出现沉淀和分层。从该实施例3-氨基丙基三乙氧基硅烷改性石墨烯的sem图中同样可以看出,还原后的石墨烯未出现团聚现象。
实施例28
3-氨基丙基三乙氧基硅烷改性氧化石墨烯的合成
将56mg氧化石墨烯加入150g四氢呋喃中,在35℃超声分散1.4h,然后加入1.34*10-2mg3-氨基丙基三乙氧基硅烷,在69℃反应7.1h,离心,洗涤,干燥,得到黑色粉末3-氨基丙基三乙氧基硅烷改性氧化石墨烯80mg。
3-氨基丙基三乙氧基硅烷改性氧化石墨烯的还原
将洗涤后烘干的20mg3-氨基丙基三乙氧基硅烷改性氧化石墨烯分散于40ml四氢呋喃中,超声0.5h后,加入0.5g水合肼,于70℃下还原6h;再用无水乙醇和蒸馏水洗涤所得产物,并烘干,得到3-氨基丙基三乙氧基硅烷改性石墨烯。称取10mg烘干后的3-氨基丙基三乙氧基硅烷改性石墨烯,分别分散于dmf,dmso,乙醇,thf,丙酮中,超声0.5~2h后,得到稳定的分散液,24h后不会出现沉淀和分层。从该实施例3-氨基丙基三乙氧基硅烷改性石墨烯的sem图中同样可以看出,还原后的石墨烯未出现团聚现象。
实施例29
3-氨基丙基三乙氧基硅烷改性氧化石墨烯的合成
将50mg氧化石墨烯加入130g四氢呋喃中,在20℃超声分散1.4h,然后加入1.01*10-2mg3-氨基丙基三乙氧基硅烷,在60℃反应7.0h,离心,洗涤,干燥,得到黑色粉末3-氨基丙基三乙氧基硅烷改性氧化石墨烯72mg。
3-氨基丙基三乙氧基硅烷改性氧化石墨烯的还原
将洗涤后烘干的20mg3-氨基丙基三乙氧基硅烷改性氧化石墨烯分散于40ml四氢呋喃中,超声0.5h后,加入0.5g水合肼,于70℃下还原6h;再用无水乙醇和蒸馏水洗涤所得产物,并烘干,得到3-氨基丙基三乙氧基硅烷改性石墨烯。称取10mg烘干后的3-氨基丙基三乙氧基硅烷改性石墨烯,分别分散于dmf,dmso,乙醇,thf,丙酮中,超声0.5~2h后,得到稳定的分散液,24h后不会出现沉淀和分层。从该实施例的3-氨基丙基三乙氧基硅烷改性石墨烯的sem图中同样可以看出,还原后的石墨烯未出现团聚现象。
实施例30
3-氨基丙基三乙氧基硅烷改性氧化石墨烯的合成
将44mg氧化石墨烯加入100g四氢呋喃中,在25℃超声分散2.0h,然后加入1.01*10-2mg3-氨基丙基三乙氧基硅烷,在66℃反应6.4h,离心,洗涤,干燥,得到黑色粉末3-氨基丙基三乙氧基硅烷改性氧化石墨烯60mg。
3-氨基丙基三乙氧基硅烷改性氧化石墨烯的还原
将洗涤后烘干的20mg3-氨基丙基三乙氧基硅烷改性氧化石墨烯分散于40ml四氢呋喃中,超声0.5h后,加入0.5g水合肼,于70℃下还原6h;再用无水乙醇和蒸馏水洗涤所得产物,并烘干,得到3-氨基丙基三乙氧基硅烷改性石墨烯。称取10mg烘干后的3-氨基丙基三乙氧基硅烷改性石墨烯,分别分散于dmf,dmso,乙醇,thf,丙酮中,超声0.5~2h后,得到稳定的分散液,24h后不会出现沉淀和分层。从该实施例的3-氨基丙基三乙氧基硅烷改性石墨烯的sem图中同样可以看出,还原后的石墨烯未出现团聚现象。
实施例31
3-氨基丙基三乙氧基硅烷改性氧化石墨烯的合成
将36mg氧化石墨烯加入89g四氢呋喃中,在21℃超声分散1.6h,然后加入8.8*10-3mg3-氨基丙基三乙氧基硅烷,在64℃反应6.4h,离心,洗涤,干燥,得到黑色粉末3-氨基丙基三乙氧基硅烷改性氧化石墨烯51mg。
3-氨基丙基三乙氧基硅烷改性氧化石墨烯的还原
将洗涤后烘干的20mg3-氨基丙基三乙氧基硅烷改性氧化石墨烯分散于40ml四氢呋喃中,超声0.5h后,加入0.5g水合肼,于70℃下还原6h;再用无水乙醇和蒸馏水洗涤所得产物,并烘干,得到3-氨基丙基三乙氧基硅烷改性石墨烯。称取10mg烘干后的3-氨基丙基三乙氧基硅烷改性石墨烯,分别分散于dmf,dmso,乙醇,thf,丙酮中,超声0.5~2h后,得到稳定的分散液,24h后不会出现沉淀和分层。从该实施例的3-氨基丙基三乙氧基硅烷改性石墨烯的sem图中同样可以看出,还原后的石墨烯未出现团聚现象。
实施例32
3-氨基丙基三乙氧基硅烷改性氧化石墨烯的合成
将32mg氧化石墨烯加入74g四氢呋喃中,在26℃超声分散1.9h,然后加入7.48*10-3mg3-氨基丙基三乙氧基硅烷,在65℃反应7.0h,离心,洗涤,干燥,得到黑色粉末3-氨基丙基三乙氧基硅烷改性氧化石墨烯44mg。
3-氨基丙基三乙氧基硅烷改性氧化石墨烯的还原
将洗涤后烘干的20mg3-氨基丙基三乙氧基硅烷改性氧化石墨烯分散于40ml四氢呋喃中,超声0.5h后,加入0.5g水合肼,于70℃下还原6h;再用无水乙醇和蒸馏水洗涤所得产物,并烘干,得到3-氨基丙基三乙氧基硅烷改性石墨烯。称取10mg烘干后的3-氨基丙基三乙氧基硅烷改性石墨烯,分别分散于dmf,dmso,乙醇,thf,丙酮中,超声0.5~2h后,得到稳定的分散液,24h后不会出现沉淀和分层。从该实施例的3-氨基丙基三乙氧基硅烷改性石墨烯的sem图中同样可以看出,还原后的石墨烯未出现团聚现象。
实施例33
3-氨基丙基三乙氧基硅烷改性氧化石墨烯的合成
将28mg氧化石墨烯加入51g四氢呋喃中,在30℃超声分散1.5h,然后加入6.17*10-3mg3-氨基丙基三乙氧基硅烷,在67℃反应6.8h,离心,洗涤,干燥,得到黑色粉末3-氨基丙基三乙氧基硅烷改性氧化石墨烯37mg。
3-氨基丙基三乙氧基硅烷改性氧化石墨烯的还原
将洗涤后烘干的20mg3-氨基丙基三乙氧基硅烷改性氧化石墨烯分散于40ml四氢呋喃中,超声0.5h后,加入0.5g水合肼,于70℃下还原6h;再用无水乙醇和蒸馏水洗涤所得产物,并烘干,得到3-氨基丙基三乙氧基硅烷改性石墨烯。称取10mg烘干后的3-氨基丙基三乙氧基硅烷改性石墨烯,分别分散于dmf,dmso,乙醇,thf,丙酮中,超声0.5~2h后,得到稳定的分散液,24h后不会出现沉淀和分层。从该实施例的3-氨基丙基三乙氧基硅烷改性石墨烯的sem图中同样可以看出,还原后的石墨烯未出现团聚现象。
实施例34
3-氨基丙基三乙氧基硅烷改性氧化石墨烯的合成
将24mg氧化石墨烯加入41g四氢呋喃中,在34℃超声分散1.1h,然后加入5.04*10-3mg3-氨基丙基三乙氧基硅烷,在64℃反应6.5h,离心,洗涤,干燥,得到黑色粉末3-氨基丙基三乙氧基硅烷改性氧化石墨烯33mg。
3-氨基丙基三乙氧基硅烷改性氧化石墨烯的还原
将洗涤后烘干的20mg3-氨基丙基三乙氧基硅烷改性氧化石墨烯分散于40ml四氢呋喃中,超声0.5h后,加入0.5g水合肼,于70℃下还原6h;再用无水乙醇和蒸馏水洗涤所得产物,并烘干,得到3-氨基丙基三乙氧基硅烷改性石墨烯。称取10mg烘干后的3-氨基丙基三乙氧基硅烷改性石墨烯,分别分散于dmf,dmso,乙醇,thf,丙酮中,超声0.5~2h后,得到稳定的分散液,24h后不会出现沉淀和分层。从该实施例的3-氨基丙基三乙氧基硅烷改性石墨烯的sem图中同样可以看出,还原后的石墨烯未出现团聚现象。
- 还没有人留言评论。精彩留言会获得点赞!