一种类石墨型氮化碳的制备方法与流程
1.本发明涉及光催化材料技术领域,具体涉及一种类石墨型氮化碳的制备方法。
背景技术:
2.工业化的快速发展和人口的迅猛增加导致全球性的资源短缺、环境污染和生态破坏,使得清洁能源和可再生能源是人类社会发展的必然选择。半导体光催化剂通过直接将太阳能转化为化学能,从而实现光解水制氢气或直接降解有机污染物,在解决能源和环境问题中表现出巨大的潜力。其中,类石墨型氮化碳,因其独特的半导体能带结构,优异的化学稳定性和良好的热稳定性等,被广泛应用于光解水制氢、传感器和光催化降解有机污染物等领域。与此同时,类石墨型氮化碳目前也在电池领域中的锂负极修饰得到进一步的应用。
3.目前,类石墨型氮化碳的制备方法主要有三种,分别是基于三聚氰胺的合成法、热解非金属硫氰化物以及剥离法。
4.例如cn111807337a公开了一种铜酞菁共轭石墨氮化碳纳米复合材料的制备方法,包括:先将三聚氰胺经加热反应制得石墨氮化碳固体,于浓硫酸中超声处理制得石墨氮化碳纳米片;然后将铜酞菁与石墨氮化碳纳米片于乙醇中超声处理,制得铜酞菁共轭石墨氮化碳纳米复合材料。其中,石墨氮化碳是利用三聚氰胺在高温下加热或煅烧得到的,虽然制备方法较为简单,但是制备出来的石墨氮化碳很难调控其孔径和比表面积。
5.cn101489923a公开了一种氮化碳的制备方法,将非金属硫氰化物简单热解以产生氮化碳材料,实现了碳和氮摩尔比为约3:4的石墨状氮化碳材料的制备。但是,所述热解非金属硫氰化物的制备方法不仅使用的原材料较为昂贵且毒性较大,而且很难对产物的孔径和比表面积进行调控。
6.cn109261207a公开了一种石墨相氮化碳的制备方法,包括:将石墨相氮化碳进行超声剥离处理得到层状石墨相氮化碳。所述剥离法首先合成体相的类石墨型氮化碳,然后利用其层间范德华力和氢键较弱的特点来剥离成片状。所述剥离法不仅使用设备或者化学手段较为复杂,而且使用原材料较为昂贵且毒性较大,还很难对产物的孔径和比表面积进行调控。
7.经文献报道,类石墨型氮化碳的光催化性能与其微观形貌(层间距、孔径、比表面积等)直接相关,而日益复杂多样的应用场合则要求类石墨型氮化碳具有不同的孔径和比表面积。因此,目前亟需开发一种新型的类石墨型氮化碳的制备方法,不仅原料简单易得、成本较低、操作简单,还可以通过调节原料的质量比例来调控类石墨型氮化碳的孔径和比表面积,便于大规模推广使用。
技术实现要素:
8.鉴于现有技术中存在的问题,本发明提供了一种新型的类石墨型氮化碳的制备方法,先将氯化铵和硫脲混合均匀,再加入去离子水中搅拌并进行水热反应,然后将固液分离
得到前驱体进行煅烧,经冷却得到类石墨型氮化碳。所述制备方法不仅可以通过调节氯化铵和硫脲的质量比,来调控类石墨型氮化碳的孔径和比表面积,使得孔径介于10
‑
200nm,比表面积介于10
‑
60m2/g,还具有原料简单易得、成本较低、操作简单等优点,便于大规模推广使用。
9.为达此目的,本发明采用以下技术方案:
10.本发明的目的在于提供一种类石墨型氮化碳的制备方法,所述制备方法包括如下步骤:
11.(1)先将氯化铵和硫脲混合均匀,再加入去离子水中搅拌并进行水热反应,然后固液分离得到前驱体;
12.(2)将步骤(1)所述前驱体进行煅烧,经冷却得到类石墨型氮化碳。
13.本发明所述制备方法以氯化铵和硫脲作为原料,一方面,先将氯化铵和硫脲混合均匀,再加入去离子水中,可以保证在进行水热反应之前,两种原料能够充分地混合均匀,进而得到均一性良好的前驱体,另一方面,氯化铵的占比是过量的,过量的氯化铵均匀地分布在前驱体的内部,在随后的高温煅烧过程中,氯化铵会分解产生nh3气体和hcl气体,气体在逸出的过程中会产生孔隙,进而可以通过调节氯化铵和硫脲的质量比,来调控类石墨型氮化碳的孔径和比表面积,使得孔径介于10
‑
200nm,比表面积介于10
‑
60m2/g,此外,所述制备方法还具有原料简单易得、成本较低、操作简单等优点,便于大规模推广使用。
14.作为本发明优选的技术方案,在步骤(1)中,氯化铵的纯度≥99.5%,硫脲的纯度≥99.5%。
15.作为本发明优选的技术方案,步骤(1)所述氯化铵和硫脲的质量比为(1
‑
4):1,例如1:1、1:1.5、1:2、1:2.5、1:3、1:3.5或1:4等,但并不仅限于所列举的数值,该数值范围内其他未列举的数值同样适用。
16.作为本发明优选的技术方案,步骤(1)所述去离子水的体积与氯化铵和硫脲质量之和的比例为(30
‑
80)ml:1g,例如30ml:1g、40ml:1g、50ml:1g、60ml:1g、70ml:1g或80ml:1g等,但并不仅限于所列举的数值,该数值范围内其他未列举的数值同样适用。
17.作为本发明优选的技术方案,步骤(1)所述水热反应的温度为40
‑
80℃,例如40℃、45℃、50℃、55℃、60℃、65℃、70℃、75℃或80℃等,但并不仅限于所列举的数值,该数值范围内其他未列举的数值同样适用。
18.优选地,步骤(1)所述水热反应的时间为2
‑
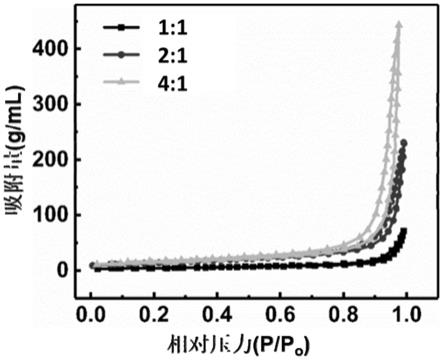
6h,例如2h、2.5h、3h、3.5h、4h、4.5h、5h、5.5h或6h等,但并不仅限于所列举的数值,该数值范围内其他未列举的数值同样适用。
19.作为本发明优选的技术方案,步骤(2)所述煅烧的温度为280
‑
330℃,例如280℃、285℃、290℃、300℃、305℃、310℃、320℃、325℃或330℃等,但并不仅限于所列举的数值,该数值范围内其他未列举的数值同样适用。
20.优选地,步骤(2)所述煅烧的时间为2
‑
6h,例如2h、2.5h、3h、3.5h、4h、4.5h、5h、5.5h或6h等,但并不仅限于所列举的数值,该数值范围内其他未列举的数值同样适用。
21.优选地,步骤(2)所述煅烧的升温速率为1
‑
10℃/min,例如1℃/min、2℃/min、4℃/min、5℃/min、7℃/min或9℃/min等,但并不仅限于所列举的数值,该数值范围内其他未列举的数值同样适用。
22.作为本发明优选的技术方案,步骤(2)所述冷却包括空冷,即利用空气进行自然冷
却。
23.作为本发明优选的技术方案,在步骤(2)所述冷却之后还包括洗涤。
24.作为本发明优选的技术方案,所述洗涤包括洗涤抽滤法。
25.优选地,所述洗涤采用的洗涤液为去离子水和/或无水乙醇。
26.作为本发明优选的技术方案,所述制备方法包括如下步骤:
27.(1)先将纯度≥99.5%氯化铵和纯度≥99.5%硫脲混合均匀,控制氯化铵和硫脲的质量比为(1
‑
4):1,再加入去离子水中搅拌,控制所述去离子水的体积与氯化铵和硫脲质量之和的比例为(30
‑
80)ml:1g,在40
‑
80℃下进行2
‑
6h的水热反应,然后固液分离得到前驱体;
28.(2)将步骤(1)所述前驱体在280
‑
330℃下进行2
‑
6h的煅烧,控制所述煅烧的升温速率为1
‑
10℃/min,将空冷得到煅烧固体采用去离子水和/或无水乙醇进行洗涤,得到类石墨型氮化碳。
29.与现有技术方案相比,本发明至少具有以下有益效果:
30.本发明所述制备方法不仅可以通过调节氯化铵和硫脲的质量比,来调控类石墨型氮化碳的孔径和比表面积,使得孔径介于10
‑
200nm,比表面积介于10
‑
60m2/g,还具有原料简单易得、成本较低、操作简单等优点,便于大规模推广使用。
附图说明
31.图1是实施例1所述类石墨型氮化碳的sem图;
32.图2是实施例1所述类石墨型氮化碳的tem图;
33.图3是实施例2所述类石墨型氮化碳的sem图;
34.图4是实施例2所述类石墨型氮化碳的tem图;
35.图5是实施例3所述类石墨型氮化碳的sem图;
36.图6是实施例3所述类石墨型氮化碳的tem图;
37.图7是实施例1
‑
3所述类石墨型氮化碳的n2吸脱附曲线。
具体实施方式
38.下面结合附图并通过具体实施方式来进一步说明本发明的技术方案。
39.为更好地说明本发明,便于理解本发明的技术方案,本发明的典型但非限制性的实施例如下:
40.实施例1
41.本实施例提供了一种类石墨型氮化碳的制备方法,所述制备方法包括如下步骤:
42.(1)先将纯度≥99.5%氯化铵和纯度≥99.5%硫脲混合均匀,控制氯化铵和硫脲的质量比为1:1,再加入去离子水中搅拌,控制所述去离子水的体积与氯化铵和硫脲质量之和的比例为50ml:1g,在50℃下进行4h的水热反应,然后固液分离得到前驱体;
43.(2)将步骤(1)所述前驱体在300℃下进行6h的煅烧,控制所述煅烧的升温速率为5℃/min,将空冷得到煅烧固体先用去离子水洗涤3次,后用无水乙醇洗涤3次,所述洗涤采用洗涤抽滤法,得到孔径为20nm,比表面积为14m2/g类石墨型氮化碳,本实施例所述类石墨型氮化碳的sem图如图1所示,tem图如图2所示。
44.实施例2
45.本实施例提供了一种类石墨型氮化碳的制备方法,除了将步骤(1)所述氯化铵和硫脲的质量比由1:1替换为2:1,其他条件和实施例1完全相同,得到孔径为40nm,比表面积为36.1m2/g类石墨型氮化碳,本实施例所述类石墨型氮化碳的sem图如图3所示,tem图如图4所示。
46.实施例3
47.本实施例提供了一种类石墨型氮化碳的制备方法,除了将步骤(1)所述氯化铵和硫脲的质量比由1:1替换为4:1,其他条件和实施例1完全相同,得到孔径为200nm,比表面积为53m2/g类石墨型氮化碳,本实施例所述类石墨型氮化碳的sem图如图5所示,tem图如图6所示。
48.实施例4
49.本实施例提供了一种类石墨型氮化碳的制备方法,除了将步骤(1)所述水热反应的温度由50℃替换为30℃,其他条件和实施例1完全相同,得到孔径为17.6nm,比表面积为12m2/g类石墨型氮化碳。
50.实施例5
51.本实施例提供了一种类石墨型氮化碳的制备方法,除了将步骤(1)所述水热反应的温度由50℃替换为100℃,其他条件和实施例1完全相同,得到孔径为22.4nm,比表面积为15m2/g类石墨型氮化碳。
52.实施例6
53.本实施例提供了一种类石墨型氮化碳的制备方法,除了将步骤(2)所述煅烧的温度由300℃替换为250℃,其他条件和实施例1完全相同,得到孔径为16.2nm,比表面积为9.7m2/g类石墨型氮化碳。
54.实施例7
55.本实施例提供了一种类石墨型氮化碳的制备方法,除了将步骤(2)所述煅烧的温度由300℃替换为350℃,其他条件和实施例1完全相同,得到孔径为24.1nm,比表面积为15.2m2/g类石墨型氮化碳。
56.实施例8
57.本实施例提供了一种类石墨型氮化碳的制备方法,所述制备方法包括如下步骤:
58.(1)先将纯度≥99.5%氯化铵和纯度≥99.5%硫脲混合均匀,控制氯化铵和硫脲的质量比为2:1,再加入去离子水中搅拌,控制所述去离子水的体积与氯化铵和硫脲质量之和的比例为50ml:1g,在40℃下进行6h的水热反应,然后固液分离得到前驱体;
59.(2)将步骤(1)所述前驱体在280℃下进行6h的煅烧,控制所述煅烧的升温速率为1℃/min,将空冷得到煅烧固体先用去离子水洗涤3次,后用无水乙醇洗涤3次,所述洗涤采用洗涤抽滤法,得到孔径为43.3nm,比表面积为37.2m2/g类石墨型氮化碳。
60.实施例9
61.本实施例提供了一种类石墨型氮化碳的制备方法,所述制备方法包括如下步骤:
62.(1)先将纯度≥99.5%氯化铵和纯度≥99.5%硫脲混合均匀,控制氯化铵和硫脲的质量比为2:1,再加入去离子水中搅拌,控制所述去离子水的体积与氯化铵和硫脲质量之和的比例为50ml:1g,在80℃下进行2h的水热反应,然后固液分离得到前驱体;
63.(2)将步骤(1)所述前驱体在330℃下进行6h的煅烧,控制所述煅烧的升温速率为10℃/min,将空冷得到煅烧固体先用去离子水洗涤3次,后用无水乙醇洗涤3次,所述洗涤采用洗涤抽滤法,得到孔径为33.5nm,比表面积为34.2m2/g类石墨型氮化碳。
64.对比例1
65.本对比例提供了一种类石墨型氮化碳的制备方法,除了将步骤(1)所述氯化铵替换为硫酸铵,其他条件和实施例1完全相同,得到孔径为7.1nm,比表面积为8.9m2/g类石墨型氮化碳。
66.对比例2
67.本对比例提供了一种类石墨型氮化碳的制备方法,除了将步骤(1)所述氯化铵替换为硫酸铵,其他条件和实施例2完全相同,得到孔径为8.3nm,比表面积为9.1m2/g类石墨型氮化碳。
68.对比例3
69.本对比例提供了一种类石墨型氮化碳的制备方法,除了将步骤(1)所述氯化铵替换为硫酸铵,其他条件和实施例3完全相同,得到孔径为8.1nm,比表面积为9.8m2/g类石墨型氮化碳。
70.上述实施例和对比例所述类石墨型氮化碳的孔径和比表面积均利用比表面积测试仪,采用n2的吸附
‑
脱附法测得;其中,将实施例1
‑
3所述类石墨型氮化碳的n2吸脱附曲线汇总在图7中,实施例1对应图例为1:1的曲线,实施例2对应图例为2:1的曲线,实施例3对应图例为4:1的曲线。
71.将实施例1
‑
3与对比例1
‑
3进行对比,可以看出采用氯化铵和硫脲作为原料,并调节氯化铵和硫脲的质量比,可以调控类石墨型氮化碳的孔径和比表面积,使得孔径介于10
‑
200nm,比表面积介于10
‑
60m2/g,如果将氯化铵替换为硫酸铵,即使调节硫酸铵和硫脲的质量比,也无法实现对类石墨型氮化碳的孔径和比表面积的调控作用,此外,所述制备方法还具有原料简单易得、成本较低、操作简单等优点,便于大规模推广使用。
72.申请人声明,本发明通过上述实施例来说明本发明的详细结构特征,但本发明并不局限于上述详细结构特征,即不意味着本发明必须依赖上述详细结构特征才能实施。所属技术领域的技术人员应该明了,对本发明的任何改进,对本发明所选用部件的等效替换以及辅助部件的增加、具体方式的选择等,均落在本发明的保护范围和公开范围之内。
73.以上详细描述了本发明的优选实施方式,但是,本发明并不限于上述实施方式中的具体细节,在本发明的技术构思范围内,可以对本发明的技术方案进行多种简单变型,这些简单变型均属于本发明的保护范围。
74.另外需要说明的是,在上述具体实施方式中所描述的各个具体技术特征,在不矛盾的情况下,可以通过任何合适的方式进行组合,为了避免不必要的重复,本发明对各种可能的组合方式不再另行说明。
75.此外,本发明的各种不同的实施方式之间也可以进行任意组合,只要其不违背本发明的思想,其同样应当视为本发明所公开的内容。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1