气凝胶及气凝胶的制造方法与流程
1.本发明涉及一种新型的气凝胶及气凝胶的制造方法,更详细而言,涉及一种包含气凝胶的新型的干燥方法的制造方法。
背景技术:
2.目前已知有一种被称作气凝胶的含有硅氧烷键的凝胶干燥体(专利文献1)。具体而言,将硅烷化合物的单体溶液(溶剂:水和/或有机溶剂)水解从而形成溶胶,使该溶胶进行缩合反应而形成凝胶(缩合化合物),然后使凝胶干燥,由此得到具有大量气孔的气凝胶(凝胶干燥体)。
3.这里气凝胶所具有的气孔的孔径为例如构成空气的元素分子在大气压时的平均自由程(mean free path[mfp])以下。因此,在气凝胶的内部几乎不会与空气进行热交换。气凝胶作为隔热材料具有优异的潜力,可以说其隔热效果仅次于真空。
[0004]
另一方面,气凝胶非常脆、难以处理、在作为制造工序的最后一道工序的干燥工序中容易因受到由制造工序中使用的水的表面张力所引起的毛细力而被破坏,因此提出了超临界干燥、冷冻干燥、通过溶剂交换的常压干燥等。
[0005]
现有技术文献
[0006]
专利文献
[0007]
专利文献1:日本专利第5250900号公报。
技术实现要素:
[0008]
发明要解决的问题
[0009]
然而,从气凝胶的成品率、大尺寸化的方面考虑,上述干燥方法还处于不能说可获得充分的生产率的状况。
[0010]
本发明的目的在于提供一种大尺寸化、具有高生产稳定性(例如高成品率)等良好的生产率和高透明性的新型的气凝胶及包含其干燥方法的气凝胶的制造方法。
[0011]
用于解决问题的方案
[0012]
本发明的目的通过下述方案来实现。
[0013]
(1)一种气凝胶,具有倍半硅氧烷结构,在含有20体积%的氧的不活性气体气氛下进行tg-dta测定时,在300~600℃的温度范围观测到两个放热峰。
[0014]
(2)根据上述(1)所述的气凝胶,其中,在观测到上述两个放热峰的温度下的分解物中,通过gc-ms测定观测到倍半硅氧烷的碎片。
[0015]
(3)一种气凝胶的制造方法,包括干燥工序,该干燥工序至少具有:
[0016]
1)第一工序,将水解物的缩合完成后的未干燥的气凝胶配置在具有第一液相和第二液相的液相体系内,
[0017]
2)第二工序,以高于室温的第一温度使构成该第一液相的第一溶剂从该第一液相中蒸发,以及
[0018]
3)第三工序,在该第一液相蒸发消失后继续保持上述第一温度来低温干燥上述未干燥的气凝胶,直到上述未干燥的气凝胶上浮至上述第二液相的液面,
[0019]
该第一溶剂为比重比构成该第二液相的第二溶剂小且沸点比构成该第二液相的第二溶剂低、并且与该气凝胶具有亲和性的溶剂。
[0020]
(4)根据上述(3)所述的气凝胶的制造方法,其中,构成上述液相体系的第一液相与气相邻接,该气相的相对湿度为50%rh以上。
[0021]
(5)根据上述(3)或(4)所述的气凝胶的制造方法,其中,在上述第三工序之后进一步具有第四工序,在上述第四工序中,将上述气凝胶从上述液相体系中取出,在比上述第一温度高的第二温度对上述气凝胶进行高温干燥。
[0022]
发明效果
[0023]
根据本发明,能够提供一种大尺寸化、具有高成品率等高生产率的新型的气凝胶及包含其干燥方法的气凝胶的制造方法。
附图说明
[0024]
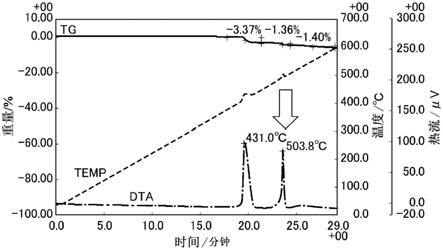
图1是本发明的气凝胶的tg-dta测定数据。
[0025]
图2是基于作为对比(专利文献1、下同)的干燥条件的气凝胶的tg-dta测定数据。
[0026]
图3是本发明的气凝胶的tg-dta测定峰的倍半硅氧烷检测gc-ms分析数据。
[0027]
图4是作为对比的气凝胶的tg-dta测定峰的倍半硅氧烷检测gc-ms分析数据。
[0028]
图5是本发明的干燥工序的示意图。
[0029]
图6是通过本发明得到的10cm
×
10cm的气凝胶。
[0030]
图7是通过本发明得到的硬币大小的气凝胶。
[0031]
图8是示出将通过本发明得到的硬币大小的气凝胶同时制造10个样品时的外观的照片。
[0032]
图9是示出在对比的条件下对与图8所示的硬币大小的气凝胶相同的组合物进行干燥时的外观照片。
具体实施方式
[0033]
以下,对本发明的实施方式进行说明。
[0034]
《气凝胶》
[0035]
本发明的气凝胶的特征在于,具有倍半硅氧烷结构,在含有20体积%的氧的不活性气体气氛下进行tg-dta测定时,在300~600℃的温度范围观测到的放热峰为两个。不活性气体优选he、n2、ar。
[0036]
《tg-dta分析》
[0037]
图1示出本发明的气凝胶的tg-dta(热重-差热同步分析)测定数据(在氦(he)气:80体积%和氧(o2)气:20体积%的混合气体中测定)。图2示出现有的作为对比的专利文献1中记载的气凝胶的tg-dta测定数据(氦(he)气:80体积%和氧(o2)气:20体积%的混合气体中)。
[0038]
在此,tg-dta(热重-差热同步分析)装置是指同时进行热重测定和差热分析测定的装置。关于图1和图2所示的三种不同的线,实线绘制的是测定热重(tg)时的重量变化“重
量/%”,虚线绘制的是以恒定的升温速度加热时的被测物质与基准物质的温度差“温度/℃”,点划线绘制的是以差热“热流/μv”表示的dta曲线。
[0039]
测定装置、测定条件如下。
[0040]
(tg―dta装置)
[0041]
差热天平rigaku thermo plus evo2(rigaku株式会社制)
[0042]
(测定条件)
[0043]
《试样量》4.370mg(图1)、4.380mg(图2)
[0044]
《气氛条件》
[0045]
·
气氛气体:80体积%的he和20体积%的氧的混合气体
[0046]
·
流量:200ml/分钟(为了抑制残留空气从天平部进入gc-ms,以300ml/分钟导入,并从天平侧以100ml/分钟排出。)
[0047]
《升温条件》温度范围:25~600℃,升温速度20℃/分钟
[0048]
《试样容器》铂(pt)
[0049]
《标准物质》in(156.6℃)、pb(327.5℃)、al(660.3℃)
[0050]
与下述gc-ms(气相色谱质谱联用仪)连接同步实施ms测定。
[0051]
(gc-ms测定装置名称)
[0052]
气相色谱质谱联用仪agilent 7890b/5977b(agilent technologies株式会社制)
[0053]
tg-dta用产生气体导入接口maff-if/d
[0054]
(测定条件)
[0055]
《离子化法》ei
[0056]
《测定法》scan
[0057]
《质量数范围》m/z:10-1000
[0058]
《gc烘箱温度》300℃
[0059]
对比图1和图2所示的tg-dta测定数据可知,本发明的气凝胶相对于对比气凝胶在高温侧具有另外的放热峰(箭头所示的峰503.8℃)。高温侧的放热峰在低温侧的放热峰+20℃~+120℃的范围被观测到。
[0060]
而且,经gc-ms确认,在该两个放热峰的温度下的分解物中包含倍半硅氧烷的碎片(m/z=252)(参照图3、图4。另外,纵轴表示相对强度。)
[0061]
此外,从图1、图2的tg数据还可知,本发明的气凝胶的重量减少与对比气凝胶相比同等或略小的程度。另外,通过比对nist库(美国国家标准与技术研究院质谱参考库)可判断出,m/z=252为倍半硅氧烷的碎片。
[0062]
由上可知,本发明的气凝胶包含对比气凝胶所不具有的热稳定性高的来自倍半硅氧烷的结构。
[0063]
虽然该来自倍半硅氧烷的热稳定性高的结构的细节尚不清楚,但是作为块体的气凝胶,其物理强度增强,结果是,使在生产上实现高成品率、大尺寸化成为可能。
[0064]
《气凝胶的内部结构》
[0065]
本发明的气凝胶除了具有tg-dta分析的结构之外,还具有下述结构。
[0066]
对于构成本发明的气凝胶的气凝胶,在微观观察其结构时,主要由充满固体物质的块体部(凝胶骨架)和以三维网眼状贯穿于块体部内的贯通孔构成,整体形成三维网络。
[0067]
另外,本发明的三维网络通过使用扫描型电子显微镜观察的状态来判断,三维网眼结构的贯通孔的直径、凝胶骨架的截面面积通过测定三维网眼状那样连续贯通孔(细孔)的中心细孔径、将骨架的截面视为圆形时的直径、进而通过压汞法测定密度、气孔率而计算。
[0068]
块体部中,固体物质由连续体构成,上述连续体基于硅氧烷键形成三维网络。三维网络中,将作为网络的最小单位的格子近似为立方体时,单边的平均长度为2nm以上且25nm以下。另外,优选单边的平均长度为2nm以上、5nm以上、7nm以上、10nm以上,并且为25nm以下、20nm以下、15nm以下。
[0069]
此外,贯通孔(细孔)呈贯穿上述块体部内的管状,当将细孔近似为管、将管的内径近似为圆时,平均内径为5nm以上且100nm以下。另外,优选细孔的平均内径为5nm以上、7nm以上、10nm以上、20nm以上、30nm以上、50nm以上,并且为100nm以下、90nm以下、80nm以下、70nm以下。在此,上述管的平均内径为构成空气的元素分子在大气压时的平均自由程(mfp)以下的尺寸。
[0070]
此外,气凝胶的气孔率、即贯通孔(细孔)的孔体积在气凝胶整体的体积中所占的比例为70%以上。作为气孔率的一个例子,可以是75%以上、80%以上、85%以上、90%以上。
[0071]
另外,本发明的气凝胶只要满足后述的物理特性,也可以包含上述的块体部、贯通孔(细孔)以外的结构。作为一个例子,也可以包含与上述的贯通孔(细孔)不同的空隙(空洞)。
[0072]
此外,作为另一个例子,只要满足后述的物理特性,还能够包含作为制造中不可避免成分而残留的水、有机溶剂、表面活性剂、催化剂以及它们的分解物。另外,作为又一个例子,只要满足后述的物理特性,还能够包含作为制造中不可避免成分而由制造空间、制造装置混入的尘埃。
[0073]
此外,本发明的气凝胶除了上述构成之外,还能够包含以赋予功能性、改善外观、赋予装饰性等为目的而添加的成分。能够包含例如抗静电剂、润滑剂、无机颜料、有机颜料、无机染料、以及有机染料。
[0074]
《气凝胶的尺寸》
[0075]
本发明的气凝胶的形状、尺寸没有限制,例如,在应用于建筑用的隔热材料等这种需要大面积的用途的情况下,优选形成具有400cm2以上的大面积的板状、片状或膜状。
[0076]
《气凝胶的密度》
[0077]
本发明的气凝胶的密度可以低至0.15g/cm3以下。在此,使用压汞法求出密度。气凝胶的密度越低,导热率越小,随之,隔热性提高。本发明的气凝胶的密度为0.15g/cm3以下,因此导热率小至0.01w/m
·
k以下。
[0078]
《气凝胶的制造:原材料》
[0079]
以下,对本发明的气凝胶及除其干燥工序以外的制造方法进行具体说明。
[0080]
《作为原材料的硅烷化合物》
[0081]
作为本发明的气凝胶所使用的硅烷化合物,优选至少将双官能硅烷化合物和三官能硅烷化合物以规定的比例(质量%)混合,尤其是将三官能硅烷化合物作为必需,由此能够赋予气凝胶柔软性。另外,通过进一步含有四官能硅烷化合物,从而能够制造裂纹等缺陷
少且密度更低的气凝胶。
[0082]
在本发明的气凝胶中,当将双官能硅烷化合物和三官能硅烷化合物的质量%分别设为dx、tx时,能够以dx∶tx=0∶100~30∶70的质量比使它们缩合,优选为5∶95~25∶75。四官能硅烷化合物优选相对于硅烷化合物整体含有0~50质量%。
[0083]
进而,硅烷化合物在双官能硅烷化合物、三官能硅烷化合物以及四官能硅烷化合物中至少包含三官能硅烷化合物,更具体而言,当将双官能硅烷化合物、三官能硅烷化合物以及四官能硅烷化合物的质量%分别设为dx、tx、qx时,至少以满足0≤dx《30、50≤tx《100、0≤qx《50、dx+tx+qx=100的比例将双官能硅烷化合物和三官能硅烷化合物混合。
[0084]
在此,双官能硅烷化合物是指硅氧烷键数量为两个的硅烷化合物,三官能硅烷化合物是指硅氧烷键数量为三个的硅烷化合物,四官能硅烷化合物是指硅氧烷键数量为四个的硅烷化合物。
[0085]
作为双官能硅烷化合物,有例如二烷氧基硅烷、二乙酰氧基硅烷。作为二烷氧基硅烷的优选的实施方式,可举出烷氧基的碳原子数为1~9的二烷氧基硅烷。具体而言,可举出二甲基二甲氧基硅烷、二乙基二甲氧基硅烷、二异丁基二甲氧基硅烷等。这些化合物可以单独使用,也可以将多个组合使用。在本发明中,作为双官能硅烷化合物,特别优选使用二甲基二甲氧基硅烷(dmdms)。
[0086]
作为三官能硅烷化合物,可举出例如三烷氧基硅烷、三乙酰氧基硅烷。作为三烷氧基硅烷的优选的实施方式,可举出烷氧基的碳原子数为1~9的三烷氧基硅烷。可举出例如甲基三甲氧基硅烷、乙基三甲氧基硅烷、丙基三甲氧基硅烷、己基三甲氧基硅烷、辛基三甲氧基硅烷、甲基三乙氧基硅烷、乙基三乙氧基硅烷、丙基三乙氧基硅烷、戊基三乙氧基硅烷、己基三乙氧基硅烷、辛基三乙氧基硅烷等。这些化合物可以单独使用,也可以将多个组合使用。在本发明中,作为三官能硅烷化合物,特别优选使用甲基三甲氧基硅烷(mtms)。
[0087]
作为四官能硅烷化合物,可举出例如四烷氧基硅烷、四乙酰氧基硅烷。作为四烷氧基硅烷的优选的实施方式,可举出烷氧基的碳原子数为1~9的四烷氧基硅烷。可举出例如四甲氧基硅烷、四乙氧基硅烷、四丙氧基硅烷、四异丙氧基硅烷等。这些硅烷化合物可以单独使用,也可以将多个组合使用。在本发明中,作为四官能硅烷化合物,特别优选使用四甲氧基硅烷(tmos)。
[0088]
《溶胶生成工序》
[0089]
用于制造本发明的气凝胶的溶胶通过包括如下溶胶生成工序的工序生成:在规定的溶液中添加包含硅烷化合物(主原料)的各种原料,进行搅拌混合。
[0090]
《溶胶生成工序的辅料和溶胶生成条件》
[0091]
在溶胶生成工序中,将双官能硅烷化合物、三官能硅烷化合物、以及任选的四官能硅烷化合物按照上述的规定混合比进行混合作为主原料,制备包含水、表面活性剂的溶液。通过该制备,硅烷化合物水解,生成含有硅氧烷键的溶胶。另外,也可以在制备的溶液中包含酸和/或有机溶剂。
[0092]
表面活性剂有利于在后述的凝胶生成工程中形成构成后述的气凝胶的块体部和气孔部。作为能够用于气凝胶的制造的表面活性剂,能够使用非离子性表面活性剂、离子性表面活性剂等。作为离子性表面活性剂,可列举出阳离子性表面活性剂、阴离子性表面活性剂、两性离子表面活性剂等。
甲基甲酰胺、n,n-二甲基甲酰胺、乙酰胺、n-甲基乙酰胺、n,n-二甲基乙酰胺等酰胺化合物、六亚甲基四胺等杂环化合物。其中,从在湿凝胶生成工序中加速生成速度的方面考虑,优选使用尿素。
[0106]
相对于100质量份的主原料的总量,碱性催化剂的添加量优选为0.5~5质量份,特别优选为1~4质量份。如果添加量小于0.5质量份,则无法进行从溶胶到湿凝胶的反应,并且,如果添加量大于5质量份,则反应过快,有时会在模具内部发生整体不均匀性。
[0107]
在它们之中,从作为催化剂的反应促进效果好、从溶胶到湿凝胶的反应时间短、且能够以低缺陷形成的方面考虑,优选氢氧化铵水溶液。此外,从挥发性高因此在后述的溶剂交换工序、干燥工序中挥发、不易残留在气凝胶中的方面考虑,也优选氢氧化铵水溶液。
[0108]
氮化物的情况下的添加量没有特别限定,例如,相对于100质量份的作为主原料的硅烷化合物的总量,优选使氮化物的添加量为1~200质量份的范围,更优选使氮化物的添加量为2~150质量份的范围。
[0109]
使添加了碱性催化剂的溶液流入到模具的工序是用于得到所期望的气凝胶产品的形状的工序。模具能够使用金属、合成树脂、木材、纸中的任一者,从兼具形状的平面性和脱模性的方面考虑,能够优选使用合成树脂。合成树脂能够例示例如聚苯乙烯、聚乙烯、聚丙烯。
[0110]
模具是用于获得所期望的气凝胶产品的形状,因此,其反映的是将所期望的气凝胶产品的形状的凹凸反转的形状。例如,在所期望的气凝胶产品的形状为板状(长方体)的情况下,能够使用一端开口的凹模盘作为模具。
[0111]
此外,模具也可以是如所谓的注射成型模具那样由多个模具形成的组合模具。作为一个例子,有凹模和凸模对置使用的两片组合模具,可以是凹模的内表面与凸模的外表面处于以规定的间隔隔离的位置关系的组合模。其结果是,溶液(由溶胶和碱性催化剂形成的溶液)可以流入到组合模具的内部空间内,在规定的时间区间内被密闭。
[0112]
此外,在使用一端开口的凹模盘作为模具的情况下,可以准备覆盖凹模盘的开放(平)面整面的平板(plate)作为第二模具,以凹模盘的开放面与第二模具对置的方式作为两片组合模具使用。其结果是,溶液(由溶胶和碱性催化剂形成的溶液)可以流入到组合模具的内部,在规定的时间区间内被密闭。
[0113]
在将添加了碱性催化剂的溶液填充至模具的工序后,具有成型工序,在该成型工序中,在模具的内部养护溶液,生成湿凝胶,并且沿着模具的内壁形状进行成型。
[0114]
养护是指施以规定的能量、经历规定的时间、推进从溶胶到湿凝胶的反应。作为能量的一个例子,为热(温度),可使用30~90℃、优选40~80℃的加热。加热可以是加热器加热,也可以是利用水或有机溶剂的蒸汽加热。
[0115]
此外,作为能量的另一个例子,可举出施加红外线、紫外线、微波、伽马射线等电磁波、施加电子射线等。这些能量可以单独使用,也可以并用多种方式。
[0116]
养护所需要的时间取决于硅烷化合物的结构、表面活性剂、水、酸、氮化物、有机溶剂、碱性催化剂等的种类和量、以及能量的种类和密度,但是在0.01小时~7天的期间。在碱性催化剂的种类和能量的种类为最优化的情况下,凝胶化可以在0.01小时~24小时内完成。此外,养护也可以是使热(温度)和时间多阶段变化的养护。另外,在湿凝胶生成及成型工序中使用的材料和/或材料的分解物可以作为不可避免的成分混入在制造的气凝胶中。
[0117]
《溶剂交换工序》
[0118]
溶剂交换工序是将湿凝胶的表面和内部所存在的水和/或有机溶剂交换成适合短时间干燥的有机溶剂的工序,如果在后续的干燥工序中需要长时间也没关系的情况下,也可以省略溶剂交换工序。此外,溶剂交换工序可以在从上述的模具取出后进行,也可以在模具内进行。
[0119]
此外,还可以在溶剂交换工序之前进行冲洗溶胶生成时使用的酸、湿凝胶生成时使用的催化剂、湿凝胶生成时使用的反应副产物等的清洗处理。清洗处理能够广泛使用有机溶剂。能够使用例如甲醇、乙醇、正丙醇、异丙醇、1-丁醇、丙酮、甲基乙基酮、甲基异丁基酮、二甲苯、1,2-二甲氧基乙烷、乙腈、己烷、甲苯、乙醚、氯仿、乙酸乙酯、四氢呋喃、二氯甲烷、n,n-二甲基甲酰胺、二甲基亚砜、乙酸、甲酸等各种有机溶剂。上述有机溶剂可以单独使用或者将两种以上混合使用。
[0120]
在它们之中,可以将对水和有机溶剂两者都具有溶解性的甲醇、乙醇、异丙醇、丙酮、甲基乙基酮等单独或两种以上混合使用。
[0121]
在溶剂交换工序中,将湿凝胶的表面和内部的水(或有机溶剂)替换为20℃时的表面张力为45mn/m以下的有机溶剂,以此来抑制在其后进行的干燥工序中的凝胶收缩损伤。可举出例如二甲基亚砜(43.5mn/m)、环己烷(25.2mn/m)、异丙醇(21mn/m)、庚烷(20.2mn/m)、戊烷(15.5mn/m)等。
[0122]
溶剂交换工序中所使用的有机溶剂的20℃时的表面张力可以为45mn/m以下、40mn/m以下、35mn/m以下、30mn/m以下、25mn/m以下、20mn/m以下、15mn/m以下,可以为5mn/m以上、10mn/m以上、15mn/m以上、20mn/m以上。在它们之中,特别是优选使用包含20℃时的表面张力为20~40mn/m范围的脂肪族烃的有机溶剂。有机溶剂能够单独使用或将两种以上混合使用。
[0123]
溶剂交换工序所使用的溶剂的量取决于溶剂交换的温度、溶剂交换的装置(容器),优选相对于湿润凝胶的容量使用2~100倍的量。溶剂交换不限于一次,可以进行多次。此外,作为溶剂交换的方法,可以是全置换、部分置换、循环置换中的任一方法。
[0124]
此外,在进行多次溶剂交换的情况下,可以针对每次溶剂交换单独设定有机溶剂的种类、温度、处理时间。另外,溶剂交换工序所使用的材料和/或材料的分解物可作为不可避免的成分混入在制造的气凝胶中。溶剂交换工序所使用的溶剂可以为构成本发明的第一液相的第一溶剂。
[0125]
《干燥工序》
[0126]
干燥工序是构成上述的本发明的气凝胶的制造方法的工序中最重要的工序。它是将进行了溶剂交换后的湿凝胶干燥而得到规定性状的气凝胶的工序,是表征本发明的气凝胶的结构的工序。
[0127]
本发明的气凝胶的制造方法的特征在于,包括干燥工序,该干燥工序至少包括:
[0128]
1)第一工序,将水解物的缩合完成后的未干燥的气凝胶配置在具有第一液相和第二液相的液相体系内,
[0129]
2)第二工序,以高于室温的第一温度使构成该第一液相的第一溶剂从该第一液相中蒸发,以及
[0130]
3)第三工序,在该第一液相蒸发消失后继续保持上述第一温度来低温干燥上述未
干燥的气凝胶,直到上述未干燥的气凝胶上浮至上述第二液相的界面上,
[0131]
该第一溶剂为比重比构成该第二液相的第二溶剂小且沸点比构成该第二液相的第二溶剂低、并且与该气凝胶具有亲和性的溶剂。
[0132]
《第一工序》
[0133]
图5中的编号1表示将水解物的缩合完成后的气凝胶(以下也称为未干燥的气凝胶)b浸渍于第一溶剂a中的状态,编号2~3相当于第一工序。
[0134]
在本发明的第一工序中,在具有第一液相d和第二液相e的液相体系内配置未干燥的气凝胶b。液相体系中形成有第一液相d和第二液相e两相,构成第一液相d的第一溶剂a与构成第二液相e的第二溶剂c选择互相完全不混合、两相分离的溶剂。第一溶剂a选择比重小于第二溶剂c的溶剂,因此形成第一液相d在上、第二液相e在下的液相体系。第一溶剂a的沸点比第二溶剂c的沸点低。
[0135]
此外,在本发明中,未干燥的气凝胶b需要具有沉降在第一液相d内进行浸渍的特性。因此,未干燥的气凝胶需要与第一溶剂具有亲和性,并且比重大于第一溶剂。
[0136]
将水解物的缩合完成后的未干燥的气凝胶b从进行缩合的反应体系取出,配置在仅充满本发明的第一溶剂a的液相体系内(图5的1)。
[0137]
本发明的第一溶剂a与气凝胶具有亲和性。在此,具有亲和性是指溶剂能够浸入到未干燥的气凝胶b的细孔中,其结果是,能够使溶剂含浸于气凝胶整体,优选例如溶解参数为7.0~9.5的溶剂。
[0138]
作为本发明的第一溶剂a,优选沸点50~100℃的有机溶剂,可举出例如:己烷(比重0.65、沸点69℃)、庚烷(比重0.68、沸点98℃)等烃系溶剂;乙酸甲酯(比重0.93、沸点57℃)、乙酸乙酯(比重0.90、沸点77℃)等酯系溶剂。
[0139]
作为本发明的第二溶剂c,只要是与第一溶剂a完全不混合能够形成d和e两个液相、且沸点高于第一溶剂a,就没有特别限制,可举出例如:水(比重1.0、沸点100℃)、硅油(比重0.94~0.98、沸点150℃以上)、氟系溶剂(比重1.3~1.5、沸点98~150℃)、汞(比重13、沸点356℃)等。
[0140]
对于未干燥的气凝胶b,例如,在气凝胶为长方体的情况下,气凝胶的底面位于第一液相d和第二液相e的大致界面处,且由于气凝胶与第一溶剂a具有亲和性,所以气凝胶的底面往上的部分大致存在于第一液相d内。
[0141]
第一液相d优选使用能够浸渍未干燥的气凝胶整体的程度的量的第一溶剂a来形成,第二液相e优选使用在第一液相d消失后能够浸渍气凝胶的大致整体的程度的量的第二溶剂c来形成。
[0142]
作为在液相体系内配置未干燥的气凝胶b的方法,如图5所示,优选采用在第一液相d内配置未干燥的气凝胶b后再添加第二溶剂c形成第二液相e的方法,但不仅限于该方法。还可以采用例如下述方法的任一方法:在形成第一液相d和第二液相e后配置未干燥的气凝胶的方法;在第二液相e上配置未干燥的气凝胶b后再添加第一溶剂a形成第一液相d的方法。
[0143]
《第二工序》
[0144]
图5中的编号4~8相当于第二工序。
[0145]
在本发明的第二工序中,以高于室温的第一温度使构成该第一液相d的第一溶剂a
从该第一液相d中蒸发。蒸发时的气氛温度优选为室温<气氛温度<沸点。此外,气氛温度可以是加热后的温度。这里所说的室温与常温相同,指15~25℃,第一温度优选比室温高的温度,优选为例如30~50℃。当第一温度在第一溶剂a的沸点以上时,气凝胶会崩解,因此不优选。
[0146]
在第二工序中,优选使存在于第一液相d之上的气相的相对湿度为50%rh以上。更优选为60%rh以上。在为了干燥而使液相体系为开放体系的情况下,优选对液相体系整体进行调湿,在将液相体系设置在干燥装置内的情况下,还能够如图5中的编号4所示那样以用通气膜f覆盖液相体系的形态形成气相。
[0147]
作为通气膜f,可举出breathron brn-3000e1、brn-9000eb、brn-1860、brn-a120e1、sunmap lc(以上为日东电工株式会社制的通气性片材、多孔膜)、细孔径为0.1~100μm的纤维素性滤纸等。
[0148]
在第二工序中,随着第一液相d消失,气凝胶在第二液相e中浸渍比例增加并且逐渐沉降(图5中的编号7),在第一液相d即将完全消失时,气凝胶几乎整体浸渍于第二液相e中(图5中的编号8)。观察到此时的气凝胶为收缩的状态,在一些情况下,可观察到向气相侧卷曲成凸状的变形。
[0149]
在第二液相e中,在卷曲的情况下,需要使用干燥中的气凝胶能够保持不与干燥装置的侧壁、底面接触的状态的量的第二溶剂c。
[0150]
第一温度在第二工序的持续期间可以进行调整变更,优选在如下时间点将加热温度稍微下调,即,在随着第一液相d被蒸发走,气凝胶从第一液相d与气相的界面露出的时间点。可举出例如在气凝胶从上述界面露出为止,将第一温度设为40℃,其后下调至30℃。
[0151]
《第三工序》
[0152]
图5中的编号9、10相当于第三工序。
[0153]
本发明的第三工序是在第一液相d蒸发消失后继续保持第一温度来低温干燥未干燥的气凝胶b、直到未干燥的气凝胶b上浮至第二液相的液面的工序。在该第三工序中,在第二工序结束的时间点,由于第一液相d消失,且由于干燥中的气凝胶的比重大于第二溶剂c的比重,干燥中的气凝胶沉降移动到第二液相e中,但是随着在第二液相e中继续蒸发,收缩的气凝胶会因回弹而溶胀恢复成长方体。
[0154]
然后,随着气凝胶内部的干燥的进行,其比重下降,当其比重小于第二溶剂c的比重时,气凝胶从第二液相e界面移动到上方。优选第三工序持续至因回弹的恢复成为稳定状态为止。将气凝胶以该状态取出,放置在大气中进行自然干燥,能够得到目标的干燥的气凝胶。
[0155]
《第四工序》
[0156]
虽然进行至本发明的第三工序就能够实现发明目的,但优选在第三工序之后还具有第四工序,在第四工序中,将气凝胶从液相体系中取出,以高于第一温度的第二温度进行高温干燥。通过进一步进行第四工序,能够得到透明性高的气凝胶。
[0157]
优选第二温度为50~250℃。在第四工序中,能够使用通常被称为恒温器的装置。例如,作为用于进行高温干燥的条件,可举出使第二温度为50~100℃、使干燥时间为5~10小时的情况,或者,使第二温度为150~250℃、使干燥时间为10~90分钟的情况。
[0158]
综上,通过经过上述的工序和干燥工序,能够抑制裂纹的产生,能够以良好的生产
率制造能够大尺寸化的透明度高的气凝胶。
[0159]
《其他工序》
[0160]
在上述的制造方法中,对呈板(或长方体)状或膜状的气凝胶的制造方法进行了说明,但本发明不限于此。作为可选工序,本发明的气凝胶的制造方法能够包括由板状的气凝胶加工出所期望的形状的工序。例如,能够由板(或长方体)加工成矩形、圆形的板或膜、正方体、球体、圆柱、棱锥、圆锥等各种形状。对于加工方法,能够使用线切割、激光切割等公知的机械加工。
[0161]
此外,作为可选工序,本发明的气凝胶的制造方法能够包括由长方体的气凝胶加工成颗粒状的气凝胶的工序。作为加工方法,能够使用颚式破碎机、辊式破碎机、球磨机等公知的粉碎机(破碎机)。
[0162]
基于这些优点,本实施方式的气凝胶能够应用于作为超低温容器、宇宙空间领域、建筑领域、汽车领域、家电领域、半导体领域、工业用设备等中的隔热材料的用途等。此外,本实施方式的气凝胶除了作为隔热材料的用途以外,还能够作为防水材料、吸音材料、防震材料、催化剂载体用等来进行利用。
[0163]
[实施例]
[0164]
以下,对本发明的实施例进行说明。
[0165]
《未干燥气凝胶的制作》
[0166]
将3.28g的作为表面活性剂的非离子性表面活性剂(basf制:pluronic pe9400)溶解在28.96g的0.005mol/l的乙酸水溶液中,进而加入4.00g的作为水解性化合物的尿素(nacalai tesque制),使其溶解。在该水溶液中添加10.00g的作为主原料的硅化合物,然后在室温搅拌混合60分钟,使硅化合物进行水解反应,生成溶胶。
[0167]
硅化合物选自作为四官能硅烷化合物的四甲氧基硅烷(多摩化学工业株式会社制的原硅酸甲酯、以下有时简称为“tmos”)、作为三官能硅烷化合物的甲基三甲氧基硅烷(dow corning toray株式会社制的dowsil z-6366silane、以下有时简称为“mtms”)、以及作为双官能硅烷化合物的二甲基二甲氧基硅烷(东京化成工业株式会社制、产品编号:d1052、以下有时简称为“dmdms”),以25质量%的四官能硅烷化合物、65质量%的三官能硅烷化合物、以及10质量%的双官能硅烷化合物的比例进行添加。另外,tmos和mtms均在使用前通过减压蒸馏进行了纯化。
[0168]
然后,使生成的溶胶在60℃在硬币大小的容器(上表面39mmφ、下表面37mmφ、高度10mm)中静置,成为凝胶。准备20个该容器。然后,继续静置96小时,由此使湿凝胶凝固。接着,从密闭容器中取出湿凝胶,将取出的湿凝胶浸渍于相当于湿凝胶体积的5倍量的甲醇(meoh)溶液中,在60℃、8小时的条件下反复进行5次溶剂交换。
[0169]
在其后的溶剂交换中,使用蒸气压高且干燥条件苛刻的己烷,将湿凝胶浸渍于以1∶4~1∶3的体积比混合异丙醇(ipa)和己烷(hex)的、相当于湿凝胶体积的5倍量的ipa/hex混合溶液中,在50℃、8小时的条件下进一步进行溶剂交换,然后,浸渍于相当于湿凝胶体积的5倍量的己烷溶液中,在50℃、8小时的条件下进一步反复进行2次溶剂交换,得到20个水解物的缩合完成后的未干燥气凝胶的样品。另外,在溶剂交换中使用的甲醇和异丙醇均由nacalai tesque制。
[0170]
《应用构成本发明的制造方法的干燥方法的实施例》
[0171]
将10个得到的未干燥气凝胶的样品如以下这样应用构成本发明的制造方法的干燥方法。该干燥方法能够在具有加热装置、顶端开放、能够倒入液体的容器中进行。
[0172]
《第一工序》
[0173]
在填充有充分覆盖整体的程度的作为构成第一液相的第一溶剂的己烷溶液的容器中放入未干燥气凝胶进行浸渍,然后向其中加入作为构成第二液相的第二溶剂的水,形成第一液相与第二液相,其中,水的加入量为己烷的同体积以上的量。在容器中,形成己烷(形成位于上侧的第一液相)与水(形成位于下侧的第二液相)分离为两个液相的液相体系,未干燥气凝胶沉降于己烷中而存在。
[0174]
《第二工序》
[0175]
在第一工序之后,使容器内的气氛温度为30℃(第一温度),蒸发己烷。此时,在开放的容器的顶面覆盖纤维素性滤纸(whatman株式会社制滤纸grade1颗粒保持能力11μm)作为通气膜,以由己烷的液面与通气膜划分出的空间的湿度为50%rh左右的方式进行调节。保持30℃左右直到己烷完全消失。
[0176]
《第三工序》
[0177]
在第二工序使己烷消失后,继续保持第一温度来低温干燥未干燥的气凝胶直到未干燥的气凝胶上浮至第二液相的液面。第二工序和第三工序的总干燥时间为48小时。在干燥过程中,未干燥气凝胶暂时在水中沉降移动,但是其后,在返回到上浮至水的液面的状态的时间点终止低温干燥,从容器取出气凝胶。
[0178]
《第四工序》
[0179]
进一步使用附加恒温器,以60℃、2小时(yamato科学株式会社制is601)、80℃、5小时(yamato科学株式会社制is601)、250℃、30分钟(yamato科学株式会社制dh412)对从容器取出的气凝胶进行再干燥,得到本发明的气凝胶。
[0180]
《采用现有的干燥方法的比较例》
[0181]
对于对比用气凝胶,将完成乙烷的溶剂交换后的10个未干燥气凝胶的样品放入到能够调节蒸发速度的容器(干燥机),在30℃干燥48小时。然后,进行与实施例的第四工序相同的再干燥,得到对比用气凝胶(比较例)。
[0182]
《评价》
[0183]
《tg-dta测定》
[0184]
测定本发明的气凝胶(实施例)和作为对比的气凝胶(比较例)的tg-dta,结果是,如图1所示,确认到实施例在300~600℃的温度范围具有两个峰。相对于此,如图2所示,确认到比较例在300~600℃的温度范围仅具有一个峰。此外,通过gc-ms测定观测到实施例中得到的两个峰均是倍半硅氧烷的碎片(图3)。
[0185]
该结果启示了,作为对比的气凝胶具有单一的倍半硅氧烷的结构,与此相对,本发明的气凝胶除了具有作为对比的气凝胶所具有的倍半硅氧烷的结构以外,还具有热稳定性更高的另一个倍半硅氧烷的结构。
[0186]
《裂纹产生观察》
[0187]
对干燥结束后的实施例、比较例的各样品进行目视观察。如图8所示,实施例中,在干燥结束的时间点,10个样品全部没有裂纹,而如图9所示,比较例中,在干燥结束时,全部的样品产生了裂纹。
[0188]
图6、图7示出了可得到透明性高、没有裂纹产生的大尺寸化的气凝胶。
[0189]
《综合评价》
[0190]
综上可知,本发明的气凝胶中抵抗干燥时的收缩力的裂纹极少,通过应用本发明的干燥方法而得到。明确可知,本发明的气凝胶的干燥方法的生产率高,还能够实现大尺寸化。
[0191]
附图标记说明
[0192]
a:第一溶剂;
[0193]
b:未干燥气凝胶;
[0194]
c:第二溶剂;
[0195]
d:第一液相(第一溶剂);
[0196]
e:第二液相(第二溶剂);
[0197]
f:通气膜;
[0198]
g:干燥完成后的气凝胶。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1