确定样本基因组中是否存在拷贝数变异的方法、系统和计算机可读介质与流程
本发明涉及确定样本基因组中是否存在拷贝数变异的方法和适于执行该方法的系统和计算机可读介质。
背景技术:
在科学研究及应用领域,时常遇到需要对单个细胞或几个细胞,或微量核酸样本进行分析的问题,比如在辅助生殖技术领域的植入前诊断(PGD)和植入前筛查(PGS),涉及对单个生殖细胞或单个卵裂球细胞或胚胎细胞进行分析;无创产前诊断技术领域,涉及通过母体外周血中微量的胎儿细胞进行检测的问题;在宏基因组学中,对环境中单个或微量的生物细胞进行分析;以及在疾病或生理研究中,涉及对组织或体液中单个细胞进行分析。
然而,目前确定拷贝数变异的方法仍有待改进。
技术实现要素:
本发明旨在至少解决现有技术中存在的技术问题之一。
根据本发明的一个方面,本发明提出了一种确定样本基因组中是否存在拷贝数变异的方法。根据本发明的实施例,该方法包括以下步骤:对所述样本基因组进行测序,以便获得由多个测序序列构成的测序结果;将所述测序结果与参照基因组序列进行比对,以便确定所述测序序列在所述参照基因组序列上的分布;基于所述测序序列在参照基因组序列上的分布,在所述参照基因组序列上确定多个突破点,所述突破点两侧的测序序列数目存在显著差异;基于所述多个突破点,在所述参照基因组上确定检验窗口;基于落入所述检验窗口的测序序列,确定第一参数;以及基于所述第一参数与预定阈值的差异,确定所述样本基因组,针对所述检验窗口是否存在拷贝数变异。利用根据本发明实施例的确定样本基因组中是否存在拷贝数变异的方法,能够有效地确定样本基因组中是否存在拷贝数变异,并且适用于各种拷贝数变异,包括但不限于染色体非整倍性、染色体片段缺失、染色体片段增加、微缺失、微重复。
根据本发明的第二方面,本发明提出了一种确定样本基因组中是否存在拷贝数变异的系统。根据本发明的实施例,该系统包括:测序装置,所述测序装置用于对对所述样本基因组进行测序,以便获得由多个测序序列构成的测序结果;分析装置,所述分析装置与所述测序装置相连,以便基于所述测序结果确定所述基因组中是否存在拷贝数变异,所述分析装置进一步包括:比对单元,所述比对单元适于将所述测序结果与参照基因组序列进行比对,以便确定所述测序序列在所述参照基因组序列上的分布;突破点确定单元,所述突破点确定单元与所述比对单元相连,并且适于基于所述测序序列在参照基因组序列上的分布,在所述参照基因组序列上确定多个突破点,所述突破点两侧的测序序列数目存在显著差异;检验窗口确定单元,所述检验窗口确定单元与所述突破点确定单元相连,并且适于基于所述多个突破点,在所述参照基因组上确定检验窗口;参数确定单元,所述参数确定单元与所述检验窗口确定单元相连,并且适于基于落入所述检验窗口的测序序列,确定第一参数;以及判断单元,所述判断单元与所述参数确定单元相连,并且适于基于所述第一参数与预定阈值的差异,确定所述样本基因组,针对所述检验窗口是否存在拷贝数变异。利用根据本发明实施例的确定样本基因组中是否存在拷贝数变异的系统,能够有效地实施根据本发明实施例的确定样本基因组中是否存在拷贝数变异的方法,从而能够有效地确定样本基因组中是否存在拷贝数变异,并且适用于各种拷贝数变异,包括但不限于染色体非整倍性、染色体片段缺失、染色体片段增加、微缺失、微重复。
根据本发明的第三方面,本发明提出了一种计算机可读介质。根据本发明的实施例,该计算机可读介质上存储有指令,所述指令适于被处理器执行以便通过下列步骤确定样本基因组中是否存在拷贝数变异:将测序结果与参照基因组序列进行比对,以便确定所述测序序列在所述参照基因组序列上的分布,其中所述测序结果是由通过对所述样本基因组进行测序所获得的多个测序序列构成的;基于所述测序序列在参照基因组序列上的分布,在所述参照基因组序列上确定多个突破点,所述突破点两侧的测序序列数目存在显著差异;基于所述多个突破点,在所述参照基因组上确定检验窗口;基于落入所述检验窗口的测序序列,确定第一参数;以及基于所述第一参数与预定阈值的差异,确定所述样本基因组,针对所述检验窗口是否存在拷贝数变异。借助该计算机可读介质,能够有效地实施根据本发明实施例的确定样本基因组中是否存在拷贝数变异的方法,从而能够有效地确定样本基因组中是否存在拷贝数变异,并且适用于各种拷贝数变异,包括但不限于染色体非整倍性、染色体片段缺失、染色体片段增加、微缺失、微重复。
本发明的附加方面和优点将在下面的描述中部分给出,部分将从下面的描述中变得明显,或通过本发明的实践了解到。
附图说明
本发明的上述和/或附加的方面和优点从结合下面附图对实施例的描述中将变得明显和容易理解,其中:
图1显示了根据本发明一个实施例的确定样本基因组中是否存在拷贝数变异的方法的流程示意图;
图2显示了根据本发明一个实施例的确定样本基因组中是否存在拷贝数变异的系统的结构示意图;
图3显示了根据本发明又一个实施例的确定样本基因组中是否存在拷贝数变异的方法的流程示意图;
图4显示了根据本发明的实施例,样品S1的染色体数字核型图。左图为胚胎单细胞经全基因组扩增后用本发明进行拷贝数变异检测的结果,右图为相同的胚胎提取DNA后直接测序(未经WGA)的结果;以及
图5显示了根据本发明的实施例,样品S2染色体数字核型图。左图为胚胎单细胞经全基因组扩增后用本发明进行拷贝数变异检测的结果,右图为相同的胚胎提取DNA后直接测序(未经WGA)的结果。
具体实施方式
下面详细描述本发明的实施例,所述实施例的示例在附图中示出,其中自始至终相同或类似的标号表示相同或类似的元件或具有相同或类似功能的元件。下面通过参考附图描述的实施例是示例性的,仅用于解释本发明,而不能理解为对本发明的限制。
需要说明的是,术语“第一”、“第二”仅用于描述目的,而不能理解为指示或暗示相对重要性或者隐含指明所指示的技术特征的数量。由此,限定有“第一”、“第二”的特征可以明示或者隐含地包括一个或者更多个该特征。进一步地,在本发明的描述中,除非另有说明,“多个”的含义是两个或两个以上。如果没有明确说明,在本文的公式或标识中,相同的字母代表相同的含义。
一、确定样本基因组中是否存在拷贝数变异的方法
根据本发明的一个方面,本发明提出了一种确定样本基因组中是否存在拷贝数变异的方法。在本发明中所使用的术语“拷贝数变异(copy number variation,CNV)”的含义是染色体或染色体片段拷贝数的异常,包括但不限于染色体非整倍性、染色体片段缺失、染色体片段增加、微缺失、微重复。
参考图1,根据本发明实施例的确定样本基因组中是否存在拷贝数变异的方法包括:
S100:对样本基因组进行测序,以便获得由多个测序序列构成的测序结果
根据本发明的实施例,本发明的方法可以采用的样本基因组的类型并不受特别限制,既可以是全基因组,也可以是基因组的一部分,例如可以是染色体或其片段。另外,根据本发明的实施例,在对样本基因组进行测序之前,进一步包括从生物样本中提取样本基因组的步骤。由此,能够直接以生物样本作为原材料,获得关于该生物样本是否具有拷贝数变异的信息,从而反映生物体的健康状态。根据本发明的实施例,可以采用的生物样本并不受特别限制。根据本发明的一些具体示例,可以采用的生物样本为选自血液、尿液、唾液、组织、生殖细胞、受精卵、卵裂球和胚胎的任意一种。本领域技术人员能够理解的是,针对不同的疾病,可以采用不同的生物样本来进行分析。由此,可以方便地从生物体获取这些样本,并且能够具体地针对某些疾病采取不同的样本,从而针对某些特殊疾病采取特定的分析手段。例如,对于可能罹患特定癌症的测试对象,可以从该组织或其附近采集样本,并进一步分离细胞进行分析,由此,能够精确并且尽可能早地获知该组织是否发生癌变。根据本发明的具体实施例,可以采用单细胞作为生物样本。根据本发明的实施例,从生物样本分离单细胞的方法和设备不受特别限制。根据本发明的一些具体示例,可以采用选自稀释法、口吸管分离法、显微操作(优选显微切割)、流式细胞分离术、微流控法的至少一种从生物样本分离单细胞。由此,能够有效便捷地获得生物样本的单细胞,以便实施后续操作,由此,可以进一步提高确定样本基因组中是否存在拷贝数变异的效率。
另外,根据本发明的实施例,对样本基因组进行测序的方法不受特别限制。根据本发明的一个实施例,对样本基因组进行测序进一步包括:首先,对样本基因组进行扩增得到经过扩增的基因组;接下来,利用经过扩增的样本基因组构建测序文库;最后,对所得到的测序文库进行测序,以便获得由多个测序序列构成的测序结果。由此,能够有效地获取样本基因组的测序结果的全基因组信息,并且能够对单细胞基因组或者微量核酸样本进行有效测序,从而进一步提高了确定样本基因组中是否存在拷贝数变异的效率。本领域技术人员可以根据采用的基因组测序技术的具体方案选择不同的构建测序文库的方法,关于构建基因组测序文库的细节,可以参见测序仪器的厂商例如Illumina公司所提供的规程,例如参见Illumina公司Multiplexing Sample Preparation Guide(Part#1005361;Feb2010)或Paired-End SamplePrep Guide(Part#1005063;Feb2010),通过参照将其并入本文。
任选地,对于以单细胞作为生物样本提取基因组,根据本发明的实施例,可以进一步包括对单细胞进行裂解,以便释放所述单细胞的全基因组的步骤。根据本发明的一些示例,可以用于裂解单细胞并释放全基因组的方法不受特别限制,只要能够将单细胞裂解优选充分裂解即可。根据本发明的具体示例,可以利用碱性裂解液将所述单细胞裂解并释放所述单细胞的全基因组。发明人发现,这样能够有效地裂解单细胞并释放出全基因组,并且所释放的全基因组在进行测序时,能够提高准确率,从而进一步提高了确定样本基因组中是否存在拷贝数变异的效率。根据本发明的实施例,单细胞全基因组扩增的方法不受特别限制,可以采用基于PCR的方法例如可以采用PEP-PCR、DOP-PCR、和OmniPlex WGA,也可以采用非基于PCR的方法例如MDA(多重链置换扩增)。根据本发明的具体示例,优选采用基于PCR的方法,例如OmniPlex WGA方法。可选用的商业化试剂盒包括但不限于Sigma Aldrich的GenomePlex,Rubicon Genomics的PicoPlex,Qiagen的REPLI-g,GEHealthcare的illustra GenomiPhi等。根据本发明的具体示例,在构建测序文库之前,可以采用OmniPlex WGA对单细胞全基因组进行扩增。由此,能够有效地对全基因组进行扩增,从而进一步提高了确定样本基因组中是否存在拷贝数变异的效率。根据本发明的实施例,可以采用选自第二代测序技术如Illumina公司的Hiseq系统,Miseq系统,Genome Analyzer(GA)系统,Roche公司的454FLX,Applied Biosystems公司的SOLiD系统,Life Technologies公司的Ion Torrent系统等的至少一种对所述全基因组测序文库进行测序。由此,能够利用这些测序装置的高通量、深度测序的特点,进一步提高了确定单细胞染色体非整倍性的效率。当然,本领域技术人员能够理解的是,还可以采用其他的测序方法和装置进行全基因组测序,例如第三代测序技术即单分子测序技术如Helicos BioSciences公司的HeliScope系统,PacBio公司的RS系统等的任一种,以及以后可能开发出来的更先进的测序技术。根据本发明的实施例,通过全基因组测序所得到的测序数据的长度不受特别限制。根据本发明的一个具体示例,所述多个测序数据的平均长度为约50bp。申请人惊奇地发现,当测序数据的平均长度为约50bp时,能够极大地方便对测序数据进行分析,提高分析效率,同时能够显著降低分析的成本。进一步提高了确定单细胞染色体非整倍性的效率,并且降低了确定单细胞染色体非整倍性的成本。这里所使用的术语“平均长度”是指各个测序数据长度数值的平均值。
S200:将测序结果与参照基因组序列进行比对,以便确定测序序列在参照基因组序列上的分布
在完成对样本基因组进行测序之后,所得到的测序结果中包含了多个测序序列。将所得到的测序结果与参照基因组序列进行比对,从而可以确定所得到的测序序列在参照基因组序列上的定位。根据本发明的实施例,可以采用任何已知的方法对这些测序数据的总数目进行计算。例如,可以采用测序仪器的制造商所提供的软件进行分析。优选采用短寡核苷酸分析包(Short Oligonucleotide Analysis Package,SOAP)和BWA比对(Burrows-Wheeler Aligner)进行,将测序序列与参考基因组序列比对,得到测序序列在参考基因组上的位置。进行序列比对可以使用程序提供的默认参数进行,或者由本领域技术人员根据需要对参数进行选择。在本发明的一个实施方案中,所采用的比对软件是SOAPaligner/soap2。
根据本发明的实施例,参照基因组序列是NCBI数据库中的标准人类基因组参考序列(例如可以为hg18,NCBI Build36)。也可以是已知基因组序列的一部分,例如可以为选自人类21号染色体、18号染色体、13号染色体、X染色体和Y染色体的至少一种的序列。
根据本发明的实施例,通过将测序结果与参照基因组序列进行比对,可以选择与参照基因组序列唯一比对的序列,进行后续分析,由此,能够避免重复序列对拷贝数变异分析的干扰,进一步提高确定样本基因组中是否存在拷贝数变异的效率。
S300:基于测序序列在参照基因组序列上的分布,在参照基因组序列上确定多个突破点
在本文中所使用的术语“突破点”指的是基因组上这样一种位点,在该位点两侧相同的区段之间测序序列数目存在显著差异。因为测序序列(reads)是来源于样本基因组的,因而当样本基因组中特定区域出现拷贝数变异时,与该区域对应的测序序列的数目也会有显著变化。由此,在确定多个突破点后,可以初步判断两个相邻突破点之间的区段可能存在拷贝数变异。
根据本发明的实施例,在参照基因组上确定突破点可以进一步包括:
首先,将参考基因组序列划分为多个预定长度的一级窗口,并确定落入各一级窗口中的测序序列。根据本发明的具体实例,可以通过常规的比对程序,将所得到的测序结果中所包含的测序序列与参照基因组序列进行比对,从而确定落入各一级窗口中的测序序列。例如可以在前面描述的S200步骤中完成。根据本发明的具体实例,落入各一级窗口中的测序序列为唯一比对测序序列。由此,能够避免重复序列对拷贝数变异分析的干扰,进一步提高确定样本基因组中是否存在拷贝数变异的效率。
接下来,针对参考基因组序列上的至少一个位点,确定落入位点两侧相同数目一级窗口中的测序序列数目。根据本发明的实施例,可以对参考基因组序列上的所有位点进行相关分析,也可以对感兴趣的染色体,例如人类21号染色体、18号染色体、13号染色体、X染色体和Y染色体的至少一种上的所有位点进行该分析。根据本发明的实施例,各一级窗口的长度可以相同或者不同,并且一级窗口之间可以有重叠,只要各个一级窗口的信息是已知的即可,优选各一级窗口具有相同的长度。根据本发明的实施例,各一级窗口的长度可以均为100-200Kbp,优选150Kbp。根据本发明的实施例,在位点两侧选择的一级窗口的数目并不受特别限制,根据具体实例,可以在位点两侧各取100个一级窗口。
接下来,可以通过统计分析,确定所研究位点两侧测序序列数据分布的p值,该p值可以反映两侧测序数据数目的显著差异性。如果所述位点的p值小于终止p值,判断该位点为突破点。根据本发明的实施例,终止p值的范围可以是通过对已知序列的样本进行平行分析而确定的,根据本发明的一个具体实例,终止p值为1.1×10-50。
根据本发明的一个实施例,确定位点两侧测序数据数目的显著差异性p值进一步包括:
针对选定的位点,在该位点两侧各取相同数目的一级窗口,并且计算每个一级窗口的相对测序序列数目Ri,其中i表示一级窗口的编号,
对所有一级窗口的相对测序序列数目Ri进行游程检验,以便确定该位点的p值,
其中,
所述相对测序序列数目是通过下列公式确定的:
其中ri表示落入第i一级窗口的测序序列数目,
n表示一级窗口的总数目。
具体地,对所有一级窗口的相对测序序列数目进行游程检验进一步包括:对每个一级窗口的相对测序序列数目Ri进行GC含量校正,以便获得校正的相对测序序列数目基于校正的相对测序序列数目,确定每个一级窗口的标准化的测序序列数目Zi;以及对所有一级窗口的标准化的测序序列数目Zi进行游程检验。更具体地,所述校正的相对测序序列数目是通过下列步骤获得的:
首先,计算每个一级窗口的GC含量;
接下来,将GC含量以预定数值为单位划分为多个区域,并且统计每个区域中相对测序序列数目的平均值Ms,其中s为GC区域的编号,根据本发明的实施例,预定数值可以为在0.0005-0.01范围内的任意数值,对应的区域大小为50k-300k,优选采用0.001,由此进行矫正的力度(power)最佳;
接下来,根据下列公式确定所述校正的相对测序序列数目
最后,通过下列公式确定标准化的测序序列数目Zi
其中,
由此,可以通过GC含量对测序序列数目进行校验。由此,可以消除基因组扩增的偏好所造成的干扰,从而进一步提高确定样本基因组中是否存在拷贝数变异的精确性和效率。
在确定多个突破点后,可以初步判断两个相邻突破点之间的区段可能存在拷贝数变异。因而可以将这些区段作为检验窗口,用于进一步判断是否存在拷贝数变异。对于初步判断得到的突破点较多的情形,可以对突破点进一步进行筛选。由此,根据本发明的实施例,基于多个突破点,在参照基因组上确定检验窗口进一步包括:
1)确定多个候选突破点,候选突破点是指这样一种突破点,在该突破点的前后均存在其他突破点;
2)确定每个候选突破点的p值,并剔除p值最大的候选突破点;
3)对剩余的候选突破点重复步骤2),在剔除一部分候选突破点,直到剩余候选突破点的p值均小于终止p值,剩余候选突破点作为经过筛选的候选突破点;以及
4)确定相邻两个经过筛选的候选突破点之间的区域为检验窗口。
根据本发明的实施例,可以通过下列步骤确定候选突破点的p值:
将候选突破点与相邻的上一个突破点之间的区域作为第一候选区域,将所述候选突破点与相邻的下一个突破点之间的区域作为第二候选区域;
对第一候选区域和第二候选区域中所包含一级窗口的标准化的测序序列数目Zi进行游程检验(游程检验是一种非参数检验,利用两个群体元素混合后的分布均匀状态评价此两个群体的差异显著性,关于该检验的细节,可以参见Wald A.WJ.On a Test Whether Two Samples are from the Same Population.The Annals of Mathematical Statistics1940;11:147-162,通过参照将其并入本文),以便确定所述候选突破点的p值。
根据本发明的实施例,终止p值是通过下列步骤确定的:
利用对照样品的测序结果,重复在参照基因组上确定检验窗口的操作,并记录每次被剔除候选突破点的p值,直到候选突破点的数目为零,这里所使用的术语对照样品指的是已知核酸序列中不存在拷贝数变异的样品;以及
基于被剔除候选突破点的p值分布,确定所述终止p值,例如将被剔除的候选突破点的p值做分布图,选取p值变化趋势最大的地方作为终止p值(pfinal)。
根据本发明的具体示例,终止p值可以为1.1×10-50。
S400:基于落入检验窗口的测序序列,确定第一参数
在确定检验窗口后,可以通过对检验窗口中所包含的测序序列进行统计分析,从而确定该检验窗口是否存在拷贝数变异。根据本发明的一个实施例,基于落入所述检验窗口的测序序列,确定第一参数进一步包括:确定检验窗口中所包含的所有一级窗口的平均标准化的测序序列数目该平均标准化的测序序列数目作为第一参数。关于标准化的测序序列数目,前面已经进行了详细描述,此处不再赘述。
S500:基于第一参数与预定阈值的差异,确定样本基因组针对该检验窗口是否存在拷贝数变异
根据本发明的实施例,可以通过将前面所确定的第一参数与预定阈值进行比较,基于第一参数和预定阈值之间的差异,来确定关于特定的检验窗口,基因组样本是否具有拷贝数变异。基于基因组测序的测序结果中,针对某一特定窗口的测序序列的数目,是与全基因组中该窗口在染色体或基因组中的含量呈正相关的,因而,通过对测序结果中来源于某一特定窗口的测序序列进行统计分析,能够有效地确定关于该窗口,样本基因组是否具有拷贝数变异。在本文中所使用的术语“预定阈值”是指将已知基因组正常的样本基因组重复针对上述实施的操作和分析所得到的关于特定窗口的相关数据。本领域技术人员能够理解的是,可以采用相同的测序条件和数学运算方法,分别获得特定窗口的相关参数,以及正常细胞的相关参数。这里,可以将正常细胞的相关参数作为预定阈值。另外,本文中所使用的术语“预定”,应做广义理解,可以是预先通过实验确定的,也可以是在进行生物样本分析时,采用平行实验获得的。这里所使用的术语“平行实验”应作广义理解,既可以指的是同时进行未知样品和已知样品的测序和分析,也可以是先后进行在相同条件下的测序和分析。根据本发明的实施例,所述预定阈值采用第一阈值和第二阈值,通过将第一参数与第一阈值和第二阈值相比较,小于第一阈值为拷贝数减少(即缺失),大于第二阈值为拷贝数增加(即重复),由此可以确定拷贝数变异类型。根据本发明的具体示例,设定α=0.05为显著界线,第一阈值为-1.645,第二阈值为1.645,由此,可以进一步有效地确定拷贝数变异类型。
利用根据本发明实施例的确定样本基因组中是否存在拷贝数变异的方法,能够有效地确定样本基因组中是否存在拷贝数变异,并且适用于各种拷贝数变异,包括但不限于染色体非整倍性、染色体片段缺失、染色体片段增加、微缺失、微重复。拷贝数变异是引起出生缺陷的主要因素,在体外培养的胚胎中也非常常见,是引起体外生殖失败的主要原因。拷贝数变异也是很多疾病如癌症的致病因素。全基因组扩增是对单个细胞、几个细胞或微量核酸样本进行全基因组范围扩增的技术,可以在尽量保持全基因组代表性前提下使样本量增加,达到所需的样本量。但是,通常而言,全基因组扩增都存在扩增偏向性的问题,有可能给后续分析带来误差。根据本发明实施例的确定样本基因组中是否存在拷贝数变异的方法,在对单细胞或微量核酸样本经全基因组扩增后,通过测序技术获得数据,进行拷贝数变异的分析,一方面通过全基因组扩增解决了单细胞或微量核酸样本分析难的问题,另一方面避免了全基因组扩增对拷贝数变异分析产生的偏差,使检测更准确更全面,尤其是通过GC含量校正,能够更进一步提高检测效率。另外,根据本发明的实施例,在不同样品的文库构建过程中引入不同的标签,由此可以同时对多种样品进行检验,进一步提高确定样本基因组中是否存在拷贝数变异的效率。利用根据本发明实施例的确定样本基因组中是否存在拷贝数变异的方法,能够进行胚胎植入前拷贝数变异筛查和诊断或无创胎儿拷贝数变异筛查,有利于提供遗传咨询和临床决策依据;进行产前诊断可有效防止病变胚胎植入,防止患儿出生。
二、确定样本基因组中是否存在拷贝数变异的系统
根据本发明的第二方面,本发明提出了一种确定样本基因组中是否存在拷贝数变异的系统,利用该系统能够有效的实施前述确定样本基因组中是否存在拷贝数变异的方法,从而可以有效的确定样本基因组中是否存在拷贝数变异。
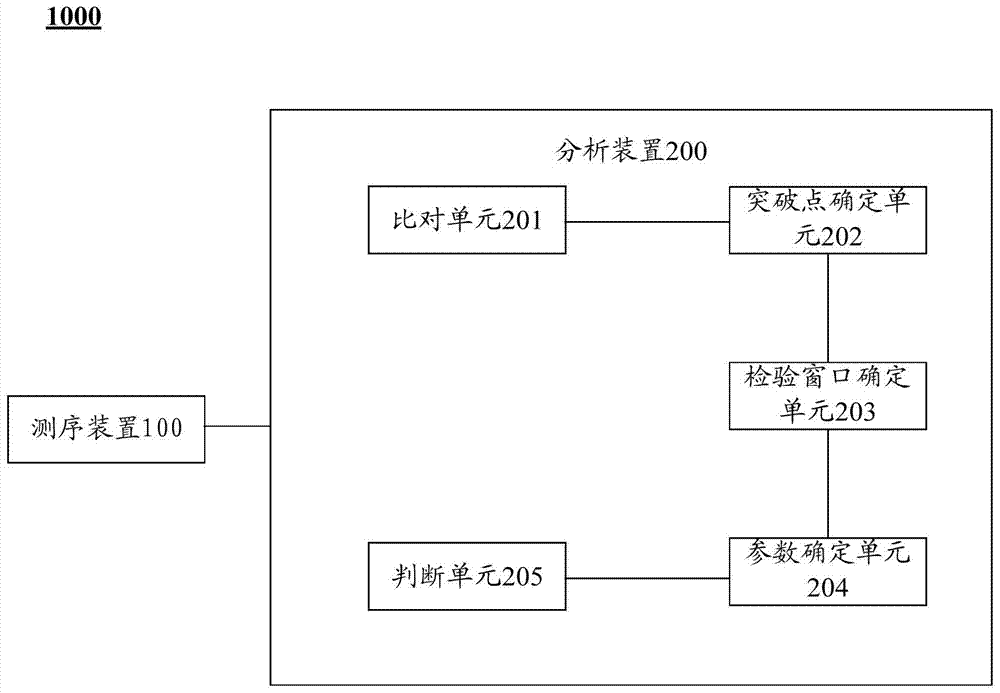
参考图2,根据本发明的实施例,确定样本基因组中是否存在拷贝数变异的系统1000包括:测序装置100和分析装置200。
根据本发明的实施例,测序装置100用于对对样本基因组进行测序,以便获得由多个测序序列构成的测序结果。根据本发明的实施例,确定样本基因组中是否存在拷贝数变异的系统1000可以进一步包括基因组提取装置(图中未示出),该基因组提取装置适于从生物样本中提取样本基因组,并且该基因组提取装置与测序装置相连以便为测序装置100提供样本基因组。由此,能够直接以生物样本作为原材料,获得关于该生物样本是否具有拷贝数变异的信息,从而反映生物体的健康状态。根据本发明的实施例,测序装置可以进一步包括:基因组扩增单元、测序文库构建单元以及测序单元。其中,基因组扩增单元适于对所述样本基因组进行扩增,测序文库构建单元与基因组扩增单元相连,并且适于利用经过扩增的样本基因组构建测序文库;以及测序单元,测序单元与所述测序文库构建单元相连,并且适于对所述测序文库进行测序。根据本发明的实施例,测序单元为选自第二代测序技术如Illumina公司的Hiseq系统,Miseq系统,Genome Analyzer(GA)系统,Roche公司的454FLX,Applied Biosystems公司的SOLiD系统,Life Technologies公司的Ion Torrent系统和单分子测序装置的至少一种。由此,能够利用这些测序装置的高通量、深度测序的特点,进一步提高了确定单细胞染色体非整倍性的效率。
根据本发明的实施例,分析装置200与测序装置100相连,以便基于测序结果确定基因组中是否存在拷贝数变异。根据本发明的实施例,分析装置200进一步包括:比对单元201、突破点确定单元202、检验窗口确定单元203、参数确定单元204以及判断单元205。其中,比对单元201适于将测序结果与参照基因组序列进行比对,以便确定测序序列在参照基因组序列上的分布。根据本发明的实施例,比对单元201内存储有参照基因组序列为已知的人类基因组序列,任选地,该参照基因组序列为选自人类21号染色体、18号染色体、13号染色体、X染色体和Y染色体的至少一种的序列。突破点确定单元202与比对单元201相连,并且适于基于测序序列在参照基因组序列上的分布,在参照基因组序列上确定多个突破点,如前所述,突破点两侧的测序序列数目存在显著差异。检验窗口确定单元203与突破点确定单元202相连,并且适于基于这些突破点,在参照基因组上确定检验窗口。参数确定单元204与检验窗口确定单元203相连,并且适于基于落入检验窗口的测序序列,确定第一参数判断单元205与参数确定单元204相连,并且适于基于所得到的第一参数与预定阈值的差异,确定样本基因组针对所确定的检验窗口是否存在拷贝数变异。
根据本发明的实施例,突破点确定单元202可以进一步包括适于执行下列以确定突破点的模块:
将参考基因组序列划分为多个预定长度的一级窗口,并确定落入各一级窗口中的测序序列;
首先,将参考基因组序列划分为多个预定长度的一级窗口,并确定落入各一级窗口中的测序序列。根据本发明的具体实例,可以通过常规的比对程序,将所得到的测序结果中所包含的测序序列与参照基因组序列进行比对,从而确定落入各一级窗口中的测序序列。根据本发明的实施例,各一级窗口的长度可以相同或者不同,并且一级窗口之间可以有重叠,只要各个一级窗口的信息是已知的即可,优选各一级窗口具有相同的长度。根据本发明的实施例,各一级窗口的长度可以均为100-200Kbp,优选150Kbp。根据本发明的实施例,在位点两侧选择的一级窗口的数目并不受特别限制,根据具体实例,可以在位点两侧各取100个一级窗口。
接下来,确定所述位点的p值,该p值可以反映两侧测序数据数目的显著差异性。以及如果所述位点的p值小于终止p值,判断该位点为突破点。根据本发明的实施例,终止p值的范围可以是通过对已知序列的样本进行平行分析而确定的,根据本发明的一个具体实例,终止p值可以为1.1×10-50。
根据本发明的实施例,突破点确定单元202可以进一步包括适于执行下列以确定p值的模块:
针对选定的位点,在该位点两侧各取相同数目的一级窗口,并且计算每个一级窗口的相对测序序列数目Ri,其中i表示一级窗口的编号
对所有一级窗口的相对测序序列数目Ri进行游程检验,以便确定所述位点的p值,
其中,
所述相对测序序列数目是通过公式确定的:
其中ri表示落入第i一级窗口的测序序列数目,
n表示一级窗口的总数目。
根据本发明的实施例,突破点确定单元202可以进一步包括适于执行下列以对所有一级窗口的相对测序序列数目进行游程检验的模块:
对每个一级窗口的相对测序序列数目Ri进行GC含量校正,以便获得校正的相对测序序列数目
基于所述校正的相对测序序列数目,确定每个一级窗口的标准化的测序序列数目Zi;以及
对所有一级窗口的标准化的测序序列数目Zi进行游程检验。其中,根据本发明的实施例,校正的相对测序序列数目是通过适于执行下列步骤的模块获得的:
计算每个一级窗口的GC含量;
将GC含量以预定数值为单位划分为多个区域,并且统计每个区域中相对测序序列数目的平均值Ms,其中s为GC区域的编号,根据本发明的实施例,预定数值可以为在0.0005-0.01范围内的任意数值,对应的区域大小为50k-300k,优选采用0.001,由此进行矫正的力度(power)最佳;
根据公式下列公式确定所述校正的相对测序序列数目
所述标准化的测序序列数目Zi是通过下列公式确定的
其中,
在确定多个突破点后,可以初步判断两个相邻突破点之间的区段可能存在拷贝数变异。因而可以将这些区段作为检验窗口,用于进一步判断是否存在拷贝数变异。对于初步判断得到的突破点较多的情形,可以对突破点进一步进行筛选。由此,根据本发明的实施例,基于多个突破点,在参照基因组上确定检验窗口进一步包括适于执行下列的模块:
1)确定多个候选突破点,候选突破点是指这样一种突破点,在该突破点的前后均存在其他突破点;
2)确定每个候选突破点的p值,并剔除p值最大的候选突破点;
3)对剩余的候选突破点重复步骤2),在剔除一部分候选突破点,直到剩余候选突破点的p值均小于终止p值,剩余候选突破点作为经过筛选的候选突破点;以及
4)确定相邻两个经过筛选的候选突破点之间的区域为检验窗口。
其中,根据本发明的实施例,通过下列步骤确定所述候选突破点的p值:
将候选突破点与相邻的上一个突破点之间的区域作为第一候选区域,将所述候选突破点与相邻的下一个突破点之间的区域作为第二候选区域;
对第一候选区域和第二候选区域中所包含一级窗口的标准化的测序序列数目Zi进行游程检验,以便确定所述候选突破点的p值。
根据本发明的实施例,终止p值是通过下列步骤确定的:
利用对照样品的测序结果,重复在参照基因组上确定检验窗口的操作,并记录每次被剔除候选突破点的p值,直到候选突破点的数目为零,这里所使用的术语对照样品指的是已知核酸序列中不存在拷贝数变异的样品;以及
基于被剔除候选突破点的p值分布,确定所述终止p值,例如将被剔除的候选突破点的p值做分布图,选取p值变化趋势最大的地方作为终止p值(pfinal)。
根据本发明的具体示例,终止p值可以为1.1×10-50。根据本发明的实施例,参数确定单元204可以进一步包括适于执行下列的模块:确定所述检验窗口中所包含的所有一级窗口的平均标准化的测序序列数目其中,所得到的平均标准化的测序序列数目作为第一参数。进一步,在判断单元205中存储有预定阙值,由此,判断单元205可以参数确定单元204所确定的第一参数与该预定阈值进行比较,从而判断所得到的检验窗口是否具有拷贝数变异,其中,根据本发明的实施例,根据本发明的实施例,所述预定阈值采用第一阈值和第二阈值,通过将第一参数与第一阈值和第二阈值相比较,小于第一阈值为拷贝数减少(即缺失),大于第二阈值为拷贝数增加(即重复),由此可以确定拷贝数变异类型。根据本发明的具体示例,设定α=0.05为显著界线,第一阈值为-1.645,第二阈值为1.645,由此,可以进一步有效地确定拷贝数变异类型。
由此,利用根据本发明实施例的确定样本基因组中是否存在拷贝数变异的系统,能够有效地实施根据本发明实施例的确定样本基因组中是否存在拷贝数变异的方法,从而能够有效地确定样本基因组中是否存在拷贝数变异,并且适用于各种拷贝数变异,包括但不限于染色体非整倍性、染色体片段缺失、染色体片段增加、微缺失、微重复。
需要说明的是,本领域技术人员能够理解,在前面所描述的确定样本基因组中是否存在拷贝数变异的方法的特征和优点也适合于确定样本基因组中是否存在拷贝数变异的系统,为描述方便,不再详述。
计算机可读介质
根据本发明的第三方面,本发明提出了一种计算机可读介质。根据本发明的实施例,该计算机可读介质上存储有指令,所述指令适于被处理器执行以便通过下列步骤确定样本基因组中是否存在拷贝数变异:将测序结果与参照基因组序列进行比对,以便确定所述测序序列在所述参照基因组序列上的分布,其中所述测序结果是由通过对所述样本基因组进行测序所获得的多个测序序列构成的;基于所述测序序列在参照基因组序列上的分布,在所述参照基因组序列上确定多个突破点,所述突破点两侧的测序序列数目存在显著差异;基于所述多个突破点,在所述参照基因组上确定检验窗口;基于落入所述检验窗口的测序序列,确定第一参数;以及基于所述第一参数与预定阈值的差异,确定所述样本基因组,针对所述检验窗口是否存在拷贝数变异。借助该计算机可读介质,能够有效地实施根据本发明实施例的确定样本基因组中是否存在拷贝数变异的方法,从而能够有效地确定样本基因组中是否存在拷贝数变异,并且适用于各种拷贝数变异,包括但不限于染色体非整倍性、染色体片段缺失、染色体片段增加、微缺失、微重复。
需要说明的是,本领域技术人员能够理解,在前面所描述的确定样本基因组中是否存在拷贝数变异的方法的特征和优点也适合于该计算机可读介质,为描述方便,不再详述。
下面将结合实施例对本发明的方案进行解释。本领域技术人员将会理解,下面的实施例仅用于说明本发明,而不应视为限定本发明的范围。实施例中未注明具体技术或条件的,按照本领域内的文献所描述的技术或条件(例如参考J.萨姆布鲁克等著,黄培堂等译的《分子克隆实验指南》,第三版,科学出版社)或者按照产品说明书进行。所用试剂或仪器未注明生产厂商者,均为可以通过市购获得的常规产品,例如可以采购自Illumina公司。
一般方法
参考图3,在实施例中采用的确定样本基因组中是否存在拷贝数变异的方法包括下列:
首先,对样品的全基因组进行扩增,并进行测序得到测序序列(测序数据);
接下来,通过SOAP2将所得到的测序序列与NCBI数据库中的标准人类基因组参考序列进行SOAP2比对,得到所测序序列在基因组上的位置信息。为避免重复序列对拷贝数变异分析的干扰,只选取与人类基因组参考序列唯一比对的测序序列(reads),进行后续分析。
接下来,寻找测试样本基因组上两侧测序序列数目在统计上有显著差异的位点,其包括下列步骤:
a)计算测试样本(可以同时对多个样本进行分析)的相对测序序列数:
在人类基因组参考序列上开长度为w的窗口(w可以是大于1的任意整数,例如10K–10M bp,优选50K–1M bp,更优选为100K–300K bp,例如约150K bp),统计所得到的测序序列中落在每个窗口上的测序序列数ri,j,其中下标i和j分别代表窗口编号和样本编号,并计算每个窗口的GC含量GCi,j,计算相对测序序列数其中平均测序序列数
b)数据校正与标准化:
在GC含量为横坐标和相对测序序列数R为纵坐标的坐标系中,将GC从小到大划分为大小相等的区域,统计每个区域中R的平均值Ms,s为GC区域的编号;
对样品中的每个窗口,计算校正的相对测序序列数窗口i的GC含量在第sGC区域内;
对于样品中每个窗口,计算标准化的相对测序序列数Zi,j:
其中
c)确定并筛选突破点
确定突破点:针对参考基因组序列上每个点,取其左右两侧各n个窗口(例如100个窗口)作为两个群体进行统计检验,每个点会计算得到一个代表该点两侧差异的p值,留下p值最小的m个点(例如3000个)作为突破点(Breakpoint)
筛选突破点:将所有排过序的突破点记为Bc={b1,b2,...,bs},每个突破点都存在左右两个片段,所述片段即上一个突破点到该突破点的区域以及该突破点到下一个突破点的区域,将这两个片段中所有Zi,j进行统计检验(例如进行游程检验,一种非参数检验,利用两个群体元素混合后的分布均匀状态评价此两个群体的差异显著性)所得的p值(pk),视作“bk作为突破点的显著性”,将pk最大的候选突破点剔除,反复此步骤,直到所有p值都小于该染色体的终止p值(pfinal);
终止p值的获得:在测试过程中,将以对照样本作为测试样本进行上述步骤确定若干突破点步骤,将全基因组上所有排过序的候选突破点记为Bc={b1,b2,...,bs},每个候选突破点bk都存在左右面两个窗口,将这两个窗口中所有Zi,j进行游程检验所得的p值(pk),视作“bk作为突破点的显著性”,将最不显著的候选突破点剔除并记录该突破点对应的p值,直到候选突破点数为0,将被剔除的候选突破点的p值做分布图,选取p值变化趋势最大的地方作为终止p值(pfinal);
确定检验窗口,并验证检验窗口:在获得经过筛选的突破点后,确定检验窗口。为了进一步对检验窗口进行判断,计算该片段中Zi,j的平均值,记为如果片段的超出阈值范围,则该片段为拷贝数变异,其中阈值的确定具体如下:
对合并窗口后每个片段,计算所有对照样品在该片段中的标准化相对测序序列数Zi,j的平均值和标准差;因为每个片段中符合正态分布,所以根据前面步骤中计算得到的平均值和标准差,计算累积概率在0.05时该片段的阈值范围,作为过滤该片段是否存在拷贝数变异的阈值。
实施例1对一例胚胎单细胞样品进行胎儿片段拷贝数变异检测,以及对一例胚胎单细胞样品进行染色体非整倍性变异检测
1、全基因组扩增:本实施例采用Sigma Aldrich公司的Single Cell Whole Genome Amplification Kit对两例胚胎单细胞样本进行全基因组扩增。所述胚胎单细胞样本为第五天囊胚期的外滋养层单细胞,通过激光捕获显微切割方法从囊胚中分离。两例胚胎单细胞经裂解后,均按照制造商所提供的试剂盒说明书进行全基因组扩增操作。
2、测序:本实施例中,采用Illumina公司的Hiseq2000测序平台对于获自上述2例胚胎单细胞全基因组扩增的DNA进行测序,按照Illumina公司所提供的说明书,进行文库构建、上机测序,使每个样本得到约0.36G数据量,每个样本根据标签序列进行区分。利用比对软件SOAP2,将测序所得测序序列与NCBI数据库中版本36(hg18;NCBIBuild36)的人类基因组参考序列进行比对,将得到测序序列在人类基因组参考序列上进行定位。
3、数据分析
a)计算测试样本和对照样本(对照样本为核型正常的样品)的相对测序序列数:
在人类基因组参考序列上划分为多个长度为150K bp的窗口,统计前面步骤2)所得到测序序列中落在每个窗口上的测序序列数ri,j,其中下标i和j分别代表窗口编号和样本编号,并计算每个窗口的GC含量GCi,j,按照一般方法中提供的公式计算相对测序序列数
b)数据校正与标准化:
在GC含量为横坐标和相对测序序列数R为纵坐标的坐标系中,将GC从小到大划分为大小为0.001的区域,统计每个区域中R的平均值Ms,s为GC区域的编号,见表1。按照一般方法中提供的公式对所得到的测序序列进行校正和标准化。
表1 校正过程中每个GC区域Ms列表
c)合并窗口
确定突破点,针对参考基因组序列上每个点,取其左右两侧各100个窗口作为两个群体进行游程检验,每个点会计算得到一个代表该点两侧差异的p值,留下p值最小的3000个点作为突破点(Breakpoint)
筛选突破点:将所有排过序的突破点记为Bc={b1,b2,...,bs},每个突破点都存在左右两个片段,所述片段即上一个突破点到该突破点的区域以及该突破点到下一个突破点的区域,将这两个片段中所有Zi,j进行游程检验所得的p值(pk),视作“bk作为突破点的显著性”,将pk最大的候选突破点剔除,反复此步骤,直到所有p值都小于该染色体的终止p值1.1×10-50;
d)在得到筛选后的突破点后,确定相邻两个突破点之间的区域为检验窗口,从而对窗口进行合并,为了进一步对合并窗口后获得的片段进行过滤,计算该片段中Zi,j的平均值,记为如果片段的超出阈值范围,则该片段为拷贝数变异。采用-1.645作为第一阈值和采用1.645作为第二阈值。
4、结果。表2表示了在该实施例中,各胚胎单细胞样品经WGA全基因组扩增后,拷贝数变异的检测结果列表。
表2.实施例1各胚胎单细胞样品经WGA拷贝数变异检测结果列表
由表2的结果,可以看出,通过本发明的确定样本基因组中是否存在拷贝数变异的方法,能够有效地确定各种类型的拷贝数变异。
实施例2
使用与实施例1相同的胚胎,重复实施例1,只是在提取DNA后直接进行测序(未经WGA)。实施例1和实施例2结果比较见表3、图4和图5。
表3.实施案例各样品WGA与未经WGA样本测序数据拷贝数变异检测结果比较
从表3数据及染色体数字核型图图4、图5可看出,样品经WGA与未经WGA测序数据拷贝数变异检测结果是一致的。对于表3“缺失”或“重复”起始终止位置(发生拷贝数变异的边界)的差异,由于拷贝数变异的边界难以精确确定,一般地,对于约150K的一级窗口,可以判定两者的边界差异在100-300Kb为完全一致,在300Kb-1Mb范围为较一致,表3显示两方法确定的拷贝数变异边界的差异范围在100-300Kb或300Kb-1Mb之内,判定两方法确定的发生拷贝数变异的边界一致。
工业实用性
本发明的确定样本基因组中是否存在拷贝数变异的方法、系统和计算机可读介质能够有效地用于确定样本基因组中是否存在拷贝数变异。
尽管本发明的具体实施方式已经得到详细的描述,本领域技术人员将会理解。根据已经公开的所有教导,可以对那些细节进行各种修改和替换,这些改变均在本发明的保护范围之内。本发明的全部范围由所附权利要求及其任何等同物给出。
在本说明书的描述中,参考术语“一个实施例”、“一些实施例”、“示意性实施例”、“示例”、“具体示例”、或“一些示例”等的描述意指结合该实施例或示例描述的具体特征、结构、材料或者特点包含于本发明的至少一个实施例或示例中。在本说明书中,对上述术语的示意性表述不一定指的是相同的实施例或示例。而且,描述的具体特征、结构、材料或者特点可以在任何的一个或多个实施例或示例中以合适的方式结合。
- 还没有人留言评论。精彩留言会获得点赞!