一种利用微生物勘探油气的方法与流程
本发明涉及微生物勘探领域,具体涉及一种利用微生物勘探油气的方法。
背景技术:
微生物勘探法是一种具有广阔前景的油气勘探方法,主要研究近地表土壤层中微生物异常与地下深部油气藏的关系。微生物勘探技术能为初期勘探(“野猫井”勘探)提供廉价有效的指示,能预测有利勘探区块以降低勘探风险。而在勘探成熟区,这项技术能将地震勘探查明的地质构造划分成各种含烃级别,并以指示油、气、水的分布位置来为油气藏开发中的油气藏表征服务。其核心在于检测和利用油气相关的微生物数量及其活性,从而判断地下油气藏的有无和分布。因此,油气指示微生物的检测技术成为整个微生物勘探法的最重要的内容之一。
待测土壤样品中微生物的数量与专一性营养物质具有正相关性,但不一定总是呈现线性相关。虽然烃氧化菌种群是决定能否氧化短链烃类的唯一因素,但烃氧化菌种群的数量却并不是影响土壤短链烃类氧化活性的唯一重要的因素。有些土样中的微生物数量较多,但活性不高;而另外一些土样中的微生物数量较低,但具有较高的活性。所以仅检测微生物菌的数量并不能准确指示地下油气分布情况,还需要检测微生物的活性。也就是说,微生物中存在不同的种类,而且真正发挥指示油气作用的微生物比较少,所以通过研究具有油气反馈功能基因的微生物可以更直接真实地反应土壤实际的原位的状况(即不但是活的,而且要是活跃的)。通常,土壤中微生物活性是利用微生物代谢产物的生化分析例如甲烷和轻烃的降解率和降解速率来评价的。现代分子生物学方法不需要培养微生物,通过系统发育以及功能基因探针的方法来直接分析土壤样品中的微生物。随着分子生物学和生物信息学的发展,加速了对微生物的认识,大量的基于核酸、脂肪酸等的分子生物学方法被应用于各种微生物的解析中,包括核酸限制性片段多样性分析、荧光原位杂交、变性梯度凝胶电泳、生物芯片、磷脂脂肪酸分析、稳定性同位素探针技术等。但由于DNA在环境中的可持留性,单纯检测样品中的DNA含量可能会造成假阳性结果的出现。另外,目前其他以细菌代谢活性或其他生理特征为基础的检测方法,操作步骤比较繁琐,检测灵敏度也不太高。
鉴于此,很有必要开发一种能够客观地评价和指示细胞的活性状态,操作简便快捷,检测灵敏度高的检测方法,以提高微生物勘探的特异性和可靠性。
技术实现要素:
本发明的目的在于提供一种提高微生物勘探的特异性和可靠性的技术,更加客观地评价和指示微生物细胞的活性状态。本发明利用mRNA具有半寿期短、非常容易被降解的特点,进行反转录定量PCR,一方面避免了由非活性菌中的功能基因(包括死菌中的功能基因或游离的功能基因)造成假阳性结果的出现;另一方面是不仅能检测到的是活性菌,而且能检测到活性菌中功能基因的拷贝数,因此而得到的相对活性数据更加能真实地反应油气藏的分布情况。
本发明中,利用反转录定量PCR技术扩增油气指示菌(油气微生物)的功能基因,检测有活性的油气微生物的数量,具有快速、灵敏、高通量、特异性强、自动化程度高、重复性好等特点。
因此,本发明提供了一种利用微生物勘探油气的方法,包括通过反转录定量PCR来定量采样点的微生物活菌中的油气功能基因拷贝数,所述采样点为目标区和包围至少1倍该目标区面积的背景区,或为目标油气区和至少1倍该目标区剖面长度上的背景区。
优选的,在本发明中,通过反转录定量PCR得到油气功能基因的mRNA的拷贝数,即微生物活菌中的功能基因拷贝数,通过定量PCR技术得到油气功能基因的DNA拷贝数,然后将油气功能基因mRNA的拷贝数/油气功能基因DNA拷贝数的值定义为微生物的相对活性,并将其作为勘探检测指标。
本发明中微生物的油气功能基因例如为甲烷氧化菌的pmoA基因、丁烷氧化菌的bmoX基因或中链烷烃氧化菌的alkB基因。当使用的油气功能基因为天然气消耗基因时,则测得的区域为气区,当使用的油气功能基因为石油消耗基因时,则测得的区域为油区。
在本发明的一个具体实施方式中,包括先将拷贝数已知的反转录定量PCR标准品进行梯度稀释,检测梯度稀释后的各个样品的反转录定量PCR的Ct值,并建立横坐标为mRNA拷贝数和纵坐标为所述Ct值的标准曲线;再使用反转录定量PCR得到待测样本中RNA反转录的Ct值后,做出标准曲线,并根据所述标准曲线计算出mRNA的拷贝数;通过定量PCR获得DNA的拷贝数。
在一个具体实施例中,比较目标区的所述油气功能基因的mRNA的拷贝数和背景区的所述油气功能基因的mRNA的拷贝数,依据统计学分析,所述目标区的mRNA的拷贝数高于背景区的mRNA的拷贝数,则将所述目标区确定为油气区。例如,在P<0.05的情况下,显著高于背景区的mRNA的拷贝数;特别是在P<0.01的情况下,显著高于背景区的mRNA的拷贝数。
在一个具体实施例中,所述目标区的mRNA的拷贝数至少高于背景区的mRNA的拷贝数的20%,优选至少高于背景区的mRNA的拷贝数的50%,特别优选至少高于背景区的mRNA的拷贝数的100%,则将所述目标区确定为油气区。
在一个具体实施例中,比较目标区的微生物的相对活性和背景区的微生物的相对活性,依据统计学分析,所述目标区的微生物的相对活性高于背景区的微生物的相对活性,则将所述目标区确定为油气区。例如,在P<0.05的情况下,显著高于背景区的微生物的相对活性;特别是在P<0.01的情况下,显著高于背景区的微生物的相对活性。
从实施例中的图4可以看出,单纯地使用mRNA的拷贝数并不能非常有效地确定油气区。而当使用微生物的相对活性时,则可以非常容易地根据测得的采样点的状况确定油气区。以图5为例,Q区的各采样点的微生物的相对活性值的平均值与B区的各采样点的微生物的相对活性值的平均值的比约为4,该比值4具有一定的参考意义,可以用于推广到真实的油气区的勘探当中。因此,在一个具体的实施例中,所述目标区的微生物的相对活性至少高于背景区的微生物的相对活性的2倍时,优选至少高于4倍时,再结合剖面整体的相对活性分布特征,可将所述目标区确定为油气区。其中的目标区的微生物的相对活性与背景区的微生物的相对活性在进行数值比较时,使用的是目标区的各个采样点的微生物的相对活性的平均值,以及背景区的各个采样点的微生物的相对活性的平均值。
本发明中的反转录定量PCR定量微生物方法是以检测靶基因的mRNA为基础的,它避免了假阳性结果的出现。尤其是以靶基因的mRNA/DNA的拷贝数比例表征土壤微生物的相对活性,因而能够更客观地评价和指示细胞的活性状态,操作快捷,检测灵敏度更高,提高了微生物勘探的特异性和可靠性。
对于微生物来说,除存在于质粒上的基因之外,存在于其染色体上的基因一般是单拷贝的,即使个别的基因为多拷贝,那么也可以通过现有技术确定其在生物体内的拷贝数。因此,当选择的基因为单拷贝数基因时,本发明测得mRNA的拷贝数可以表示为有活性的微生物的数量;DNA的拷贝数则可以表示为有活性和无活性的微生物的数量的总和。即使选择的基因为多拷贝数基因,也可以通过将DNA的拷贝数换算后得到微生物的数量的总和。
本发明还提供了反转录定量PCR方法在油气勘探中表征微生物相对活性的应用。
本发明进一步提供了油气功能基因的mRNA的拷贝数在油气勘探中表征微生物相对活性的应用。
在本发明中,对于采样点的选择以及采样点深度的选择,是本领域技术人员公知的。也就是说,本领域的技术人员可以根据目标区的介质均匀度及土壤湿度等状况来确定采样点和采样深度。
本发明中反转录定量PCR技术定量微生物的优点至少体现在如下几个方面:
1)该检测技术具有快速、灵敏的特点,并可适合于批量化高通量的基因定量检测;进行大规模土壤样品的微生物检测时能够大幅度缩短时间,提高效率。
2)mRNA具有半寿期短,非常容易被降解的特点,以检测mRNA为基础的反转录定 量PCR(RT-qPCR)能够客观地评价和指示细胞的活性状态,避免了单纯检测样品中的DNA含量可能造成假阳性结果的现象。
3)若以mRNA/DNA的功能基因的拷贝数比例表征土壤微生物的相对活性,加大了不同区域的非活性菌与活性菌数据之间的差别,更加完善了微生物勘探检测指标。
附图说明
图1为pmoA基因定量PCR荧光曲线。
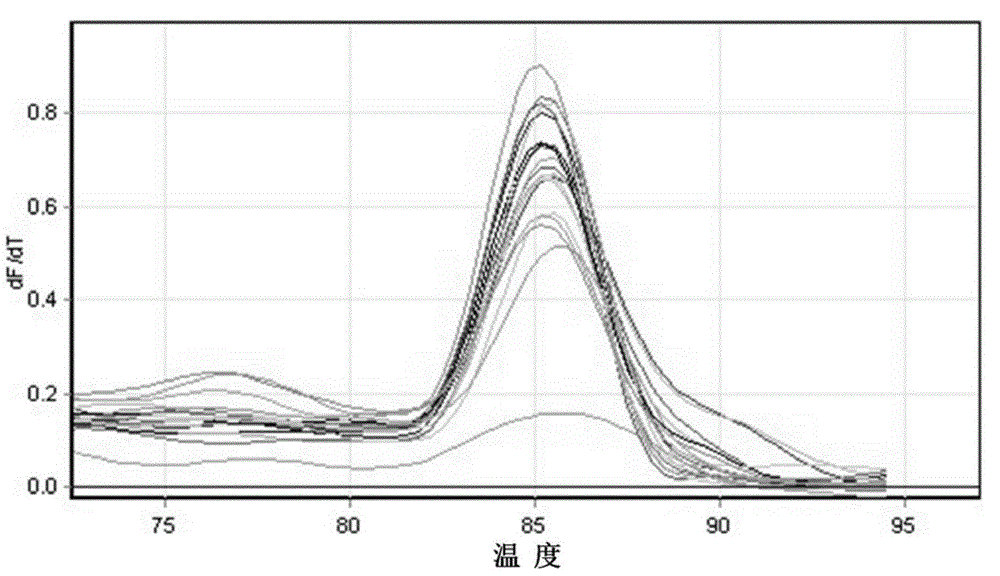
图2为pmoA基因定量PCR溶解曲线。
图3为pmoA基因定量PCR产物的电泳图谱。其中,M为DNA碱基对个数参照物DL2000;1:原样;2-9:10-1-10-8稀释梯度;N:空白对照(NTC)。
图4为某区块B剖面30cm深处RNA反转录定量结果。
图5为RNA反转录定量PCR得到的拷贝数占DNA定量PCR得到的拷贝数的比值数据结果。
图4和图5中的横坐标均为不同的样本采集点,具体包括B01-B14、Y14-Y33、B21-B28、Q42-Q81和B37-B43中的多个样本采集点;而图4的纵坐标为mRNA的拷贝数/g干土,图5的纵坐标为相对活性数据。
具体实施方式
在以下实施例中,微生物的油气功能基因以甲烷氧化菌的pmoA基因为例。
本发明中,相对活性=土壤中微生物活菌中的油气功能基因的拷贝数/土壤中总功能基因(包括活菌中的、死菌中的和游离的)的拷贝数,或者是相对活性=mRNA的拷贝数/DNA功能基因的拷贝数。
实施例1
mRNA的拷贝数和DNA拷贝数的计算
本实施例提供一种反转录定量PCR标准品(一种mRNA)的构建,该标准品是在DNA模板和RNA合成酶催化下经转录得到。根据该标准品的不同浓度梯度得到相应的定量PCR的循环数Ct值,并作出以横坐标为mRNA的拷贝数/微升和纵坐标为mRNA的Ct值的标准曲线。
[1]目的基因扩增
PCR反应(20μL):2μL 10×PCR缓冲液、1.6μL d-NTPs(200mM)、0.3μL Taq-HS酶(1.5U)、1μL正向引物A189f(10mM);、1μL反向引物mb661r(10mM)、13.7μL去离子水、0.4μL DNA模板;
其中,DNA模板是从野外采集的土壤环境样品获得,目的是为了最终获得pmoA基因。
PCR的反应程序:94℃变性阶段10min,94℃变性50s,60℃退火50s,72℃延 伸40s,共35个循环,循环结束后72℃充分延伸10min。
[2]PCR产物的片段纯化
使用试剂盒TaKaRa Agarose Gel DNA Purifcation Kit Ver.2.0,方法参照说明书。
[3]连接反应
用pGEM-T Easy载体连接上述PCR产物,用于转入大肠杆菌JM109中,反应体系(10μL)为5μL T4缓冲液、1μL pGEM-T Easy载体(50ng)、3μL PCR产物、1μL T4连接酶(3weiss units/μL)。反应条件:4℃过夜充分反应。
[4]PCR产物的转化
将制备好的感受态大肠杆菌JM109从-70℃取出,置冰上融化,加入冰预冷的上述连接反应产物5μL,轻轻混匀;冰浴20min,转到42℃水浴中热激90s,随后快速冰浴2min;加入950mL SOC培养基,混匀,置于37℃,150rpm培养1.5h。同时在预先倒好的含100μg/mL氨苄青霉素的LB平板上涂布30μL的IPTG(40mg/mL)和40μL的X-gal(20mg/mL),37℃倒置平皿,使其被培养基充分吸收。将培养1.5h的菌液取出,涂布200μL于准备好的平板,37℃培养12-14h培养。
[5]蓝白斑筛选
用枪头随机挑取白色的菌落,转移至20mL含100μg/mL氨苄青霉素的LB液体培养基中,37℃摇床过夜。所得菌液一部分送样测序。
[6]提取的质粒EcoR I酶切验证图谱并测序
反应体系(20μL):2μL 10×H缓冲液、1μL EcoR I酶、质粒DNA 4μL、ddH2O补齐20μL。反应条件:37℃反应180min后65℃反应10min灭活EcoR I酶活性。将携带该克隆子的E.Coli JM109进行质粒测序,测序后的序列上传NCBI后得到的基因银行(Gene Bank)序列号为JN106047。
[7]对提取的质粒在插入片段进行下游单酶切
反应体系(20μL):2μL 10×M缓冲液、1μL Spe I酶、质粒DNA 4μL、无核糖核酸酶H2O补齐20μL。反应条件:37℃反应180min后65℃反应10min灭活Spe I酶活性。
[8]体外转录合成RNA
反应体系(20μL):2μL 10×T7RNA聚合酶缓冲液、2μL DTT(50mM)、2μL NTP混合物(四种NTP中的每种的浓度为2.5mM)、0.5μL核糖核酸酶抑制剂(40U/μL)、质粒DNA 10μL、T7RNA聚合酶10-50U、无核糖核酸酶H2O补齐20μL。反应条件:37℃反应60min。
[9]消化体外转录合成的RNA样品中的模板DNA
向上步得到的RNA溶液中加入10μL含有MgCl2的10×反应缓冲液、10μL DNase I(无核糖核酸酶)、无核糖核酸酶的H2O补齐100μL;37℃反应30min后加入10μL EDTA (50mM)以防止加热过程中RNA的水解,65℃反应10min灭活DNase I活性;加入1/10体积的NaAc(3M)和2.5倍体积的无水乙醇,混匀后-20℃放置30min以沉淀RNA;4℃条件下以12,000rpm转速离心10min后弃掉上清液,得到的沉淀用100μL无核糖核酸酶水重新溶解沉淀,即为反转录定量PCR的标准品(mRNA)。
[10]建立反转录定量PCR标准曲线方法
测定转录得到的mRNA(该标准品)的OD值,计算拷贝数后,将mRNA缓冲液进行10倍梯度稀释,进行反转录定量PCR,反转录定量PCR的荧光曲线图,得到的曲线图如图1所示。根据图1对引物进行定量PCR的可行性分析,结果显示完成45个循环扩增后,观察荧光扩增曲线,曲线呈现良好的“S”形。并测定pmoA基因的PCR溶解曲线(见图2),结果表明,融解曲线在85℃出现单峰,说明定量PCR扩增具有较强的特异性。对不同稀释梯度的DNA样品定量PCR产物进行凝胶电泳(见图3)分析得出,扩增得到了清晰的目标条带,无非特异性扩增现象的出现,说明扩增pmoA基因的PCR条件较佳。然后对反转录定量PCR进行核酸电泳检测(见图3)。并构建反转录定量PCR标准曲线(见图4)。拷贝数(其单位为bp)的计算公式为:
根据标准曲线建立方程式:y=ax+b,其中x为阈值(Threshold value)(拷贝数/微升),y为拷贝数的对数值;a为斜率;b为截距。本发明中的标准曲线为:计算标准曲线为y=-3.281x+34.006,其R2达到了0.99以上,符合检测标准。检测范围为102-109拷贝数。
使用本实施例中得到的标准曲线可用于检测土壤中微生物活菌中的功能基因的拷贝数,即mRNA的拷贝数。具体的,先检测土壤中微生物活菌中的功能基因的反转录定量PCR的Ct值,再根据该标准曲线读出相应的拷贝数值。另外,本领域技术人员可知,对于微生物中的同一个功能基因,仅需做一次反转录定量PCR标准曲线即可。
DNA拷贝数的计算方法参考mRNA拷贝数的计算方法,不同之处在于,在此过程中不存在反转录的步骤。
实施例2
土壤样品检测方法
首先按照如下步骤A-G提取环境样品中的总RNA,再采用反转录定量PCR测定样品Ct值,基于实施例1中获取的反转录定量PCR标准曲线,得出微生物活菌中功能基因的拷贝数。再根据常规的DNA提取方法获得土壤中的总DNA,使用定量PCR检测后得到DNA的拷贝数(土壤样品中总的功能基因的拷贝数)。在获得mRNA和DNA的数据以后,计算mRNA/DNA的比值(见图5),并以mRNA/DNA的比例表征土壤微生物的相对活性。
其中,提取土壤样品中的总RNA的步骤如下:
A.称取0.5g土壤至2mL离心管中,加入0.5g玻璃珠(直径0.5mm),0.5mL苯酚:氯仿:异戊醇(25:24:1),0.5mL CTAB提取液(5%CTAB,2mol/L NaCl,0.12mol/L磷酸钠缓冲液pH 8.0);
B.至涡旋仪上以最大转速振荡10min;
C.12,000rpm,4℃离心10min;
D.加入等体积的氯仿:异戊醇(24:1),12,000rpm,4℃离心10min,重复一次;
E.加入0.6倍体积的异丙醇-20℃沉淀1h;
E'.加入2倍体积的30%PEG-1.6mol/L NaCl室温沉淀2h;
E”.加入1/10体积的3mol/L NaAc,2.5倍体积的无水乙醇沉淀1h;
F.14,000rpm,4℃离心20min;
G.加入50μL DEPC处理水溶解沉淀。取5μL RNA样品,用1%琼脂糖电泳检测完整性,紫外分光光度计(Nanodrop ND2000)测定RNA溶液浓度,记录紫外A230、A260、A280处的吸收值,并用A260/A230和A260/A280鉴定纯度。
实施例3
准确性的验证
选择已知的包含油气区和背景区的剖面按方向顺序取样(采样深度为60cm),然后按照本实施例中的方法在某油气区进行微生物勘探的验证性研究,使用本发明所述的反转录定量PCR技术定量微生物,图4中显示的是某油气区土壤样品提取得到的RNA经过反转录定量PCR后的结果,其结果显示,各样品的拷贝数约分布在103-104拷贝数/g干土的范围内,一些RNA样品结果在最低检测线以下,经电泳检测未见目的条带,此方法在气区(Q区)显示出一定异常高值,背景区(B区)显示出较弱的高值,这是因为背景区油气逸散较少或者没有,造成了以油气为食物的微生物菌无法广泛发育。油区(Y区)则显示出异常的低。图5显示的是RNA反转录定量PCR得到的拷贝数占DNA定量PCR得到的拷贝数的比例,从图5中可以看出在气区出现的是连片的高活性异常值,且边界清晰,说明该指标可以丰富油气藏指示,以使其更好地应用于实际。通过图4和图5的比较可以直观的看出,由于图4仅使用mRNA的拷贝数来确定油气区,各样品的mRNA的拷贝数约分布在103-104拷贝数/g干土的范围的区域有两个,一个是背景区的一部分,另一个是真正的气区;而由于图5中的各个样品的数值是mRNA的拷贝数/DNA拷贝数的比值,则将背景区与气区区分开来,从而避免的假阳性的产生,因此,相比于仅使用mRNA的拷贝数作为判断指标来看,以mRNA的拷贝数/DNA拷贝数作为判断指标具有更高的可靠性。但是两个的可靠性均高于DNA的拷贝数作为判断指标。
- 还没有人留言评论。精彩留言会获得点赞!