(R)-拉多替吉的R型手性中间体和类似物的制备及其应用的制作方法
本发明涉及化学物质和药物制备领域,涉及多靶点治疗轻度认知功能障碍药物(R)-拉多替吉的R型手性中间体和类似物的制备及其应用。
背景技术:
动态动力学拆分(Dynamic Kinetic resolution,DKR)是合成手性胺和醇等光学纯异构体的新方法,其过程包括动力学拆分和底物外消旋化反应,是在对映体选择性转化的同时进行酶或非酶催化的原位消旋化,从而使消旋的起始原料更多地转化为单一对映体。DKR过程理论上可使底物转化率接近100%,产物ee值达到98%以上。
拉多替吉(Ladostigil)是一种新型的治疗轻度认知功能障碍(MCI)的候选药物,该药物具有一个手性中心,其R型异构体的活性和安全性都远远好于S型异构体。拉多替吉最初被设计为综合AD治疗药物——卡巴拉汀(Rivastigmine)与PD治疗药物——雷沙吉兰的药效团,大剂量下通过抑制乙酰胆碱酯酶和脑内单胺氧化酶(MAO)的活性,用于治疗AD。该药由以色列Teva公司研发,在投资数千万美元开发和测试后被耶路撒冷希伯来大学的Yissum公司收购,亚伯拉罕制药公司又从Yissum购买了该药的专利,对其作了一些修改,并恢复了用其治疗AD的试验。2012年底结束的临床Ⅱb期试验结果表明未达到预期目标,但实验证实小剂量的拉多替吉具有很好的神经保护作用。
目前拉多替吉的合成路线主要为以下三条:
1. Youdim M.B等首次报道的路线,采用(R)-1-氨基茚满为原料,成本较高,反应收率较低,造成手性原料的很大浪费。
2. Nudelman等报道的路线,较简单,但得到的产物为消旋体,需拆分出R型对映体,不够经济。
3.专利US20060199974(A1)描述的路线,关键步骤采用Ru-TsDPEN催化氢转移反应,将酮转变为手性醇,催化剂用量大,成本高,无实际利用价值。黎星术等对此法进行改进,关键步骤采用Ru–Cs-DPEN催化剂在含表面活性剂的甲酸钠-水体系中进行,收率和ee值均大于99%,催化剂可以回收5次,但实验中用到的炔丙胺毒性很大,催化剂成本依然高昂,该法仅适用于实验室小量制备。
技术实现要素:
本发明所解决的技术问题是提供一种制备(R)-拉多替吉的R型手性中间体和类似物的方法,还提供了所制备中间体在制备(R)-拉多替吉中的应用。
本发明是通过如下技术方案实现的:
本发明所述的(R)-拉多替吉的R型手性中间体和类似物(即化合物4)通过如下方法制备:
底物化合物3与酰基供体7在碱性条件下,在钯催化剂与酶的催化下通过动态动力学拆分(DKR)反应得到化合物4;
R1为H,C1~C10烷基,C2~C10烯基,C2~C10酮,C2~C10酯;
R2为C1~C10烷基,C2~C10烯基、C6~C10芳基、含1-3个氮或硫的6-10元芳香杂环;
R3为C1~C10烷基、C1~C10烷氧基、C2~C10烯基、C6~C10芳基、或含1-3个N,O或S的杂原子的5-10元杂环,所述烷氧基可以被C1~C10烷基取代。
优选地,
R1为H,C1~C4烷基;
R2为C1~C4烷基;
R3为C1~C4烷基、C1~C4烷氧基、所述烷氧基可以被C1~C4烷基取代。
所述钯催化剂为Pd/C,Pd/BaSO4,Pd/CaCO3,Pd/BaCO3,Pd/AlO(OH)。
所述的酶为南极假丝酵母脂肪酶A或B(Candida antarctica lipase A,Candida antarctica lipase B,CALA或CALB)。
所述的碱为无水碳酸钠,碳酸氢钠,碳酸钾等。
反应温度20~80℃;酶与底物的用量比为1:1~1:10;反应搅拌方式为非直接接触酶的混匀方式;底物与酰基供体的用量比为1:1~1:5;底物容量为10g/L~200g/L;反应时间从1h~24h,反应溶剂主要包括甲苯,正己烷,氯仿,异丙醚,乙二醇,二甲醚,四氢呋喃,二氯甲烷,乙腈,丙酮,乙醚。
化合物3可以通过如下反应制备得到:
化合物2在还原剂的条件下被还原成化合物3,所述的还原剂为:硼氢化钠,四氢铝锂,硼烷,氢化铝锂,亚硫酸钠,亚硫酸氢钠,优选NaBH4。
化合物2可以通过如下反应得到:
将化合物1用乙醇水溶液溶解,加入盐酸羟胺,后用氢氧化钠处理即得到化合物2。
进一步地,本发明制备的化合物4可以用于制备(R)-拉多替吉。
所述的(R)-拉多替吉的制备方法如下:
(1)以化合物1为原料,在盐酸羟胺和氢氧化钠的作用下得到化合物2;
(2)化合物2在还原剂的作用下得到化合物3;
(3)化合物3与酰基供体在碱性条件下,在钯催化剂与酶的催化下通过动态动力学拆分(DKR)反应得到化合物4;
(4)化合物4通过常规制备方法得到化合物5。
(5)化合物5通过常规制备方法得到化合物6((R)-拉多替吉)。
R1位H,C1~C10烷基,C2~C10烯基,C2~C10酮,C2~C10酯;
R2为C1~C10烷基,C2~C10烯基、C6~C10芳基、含1-3个氮或硫的6-10元芳香杂环;
R3为C1~C10烷基、C1~C10烷氧基、C2~C10烯基、C6~C10芳基、或含1-3个N,O,S的杂原子的5-10元杂环,所述烷氧基可以被C1~C10烷基取代。
优选地,
R1为H,C1~C4烷基;
R3为C1~C10烷基、C1~C4烷氧基、所述烷氧基可以被C1~C4烷基取代。
所述的反应中,
其中,步骤(1)中化合物通过如下反应得到:
将化合物1用乙醇水溶液溶解,加入盐酸羟胺,后用氢氧化钠处理即得到化合物2。
步骤(2)中化合物3通过如下反应制备得到:
化合物2在还原剂的条件下被还原成化合物3,所述的还原剂为:硼氢化钠,四氢铝锂,硼烷,氢化铝锂,亚硫酸钠,亚硫酸氢钠,优选NaBH4。
化合物2中6位取代基的供电子共轭效应使其C=N键电子云密度增高,导致其不易被还原。发明用NiCl2增效的NaBH4还原,最终得到化合物3。
步骤(3)中所述化合物4通过如下反应制备得到:
所述钯催化剂为Pd/C,Pd/BaSO4,Pd/CaCO3,Pd/BaCO3,Pd/AlO(OH)。
所述的酶为南极假丝酵母脂肪酶A或B(Candida antarctica lipase A,Candida antarctica lipase B)。
所述的碱为无水碳酸钠,碳酸氢钠,碳酸钾等。
反应温度20~80℃;酶与底物的用量比为1:1~1:10;反应搅拌方式为非直接接触酶的混匀方式;底物与酰基供体的用量比为1:1~1:5;底物容量为10g/L~200g/L;反应时间从1h~24h,反应溶剂主要包括甲苯,正己烷,氯仿,异丙醚,乙二醇,二甲醚,四氢呋喃,二氯甲烷,乙腈,丙酮,乙醚。
本发明中,我们通过动态动力学拆分(DKR)反应高产率的得到了R型手性中间体(化合物4),并将其应用于合成(R)-拉多替吉,为探索一条绿色经济的拉多替吉合成路线打下基础。从而提高了反应效率,简化了工艺。
附图说明
图1是R1为甲基,R3为甲基甲氧基时进行DKR反应所得到的产物4的红外图谱。
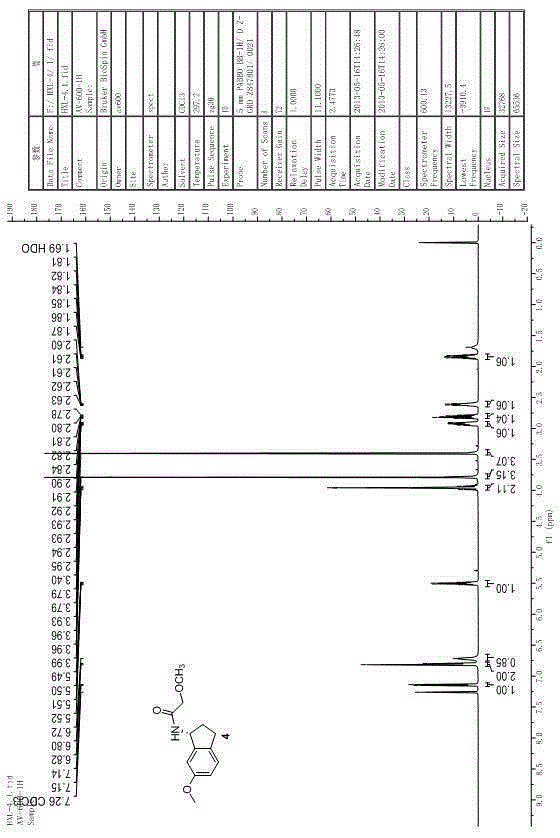
图2是R1为甲基,R3为甲基甲氧基时进行DKR反应所得到的产物4的核磁图谱。
图3是R1为甲基,R3为甲基甲氧基时进行DKR反应所得到的产物4的液相图谱。
具体实施方式
实施实例1:
化合物2(R1=CH3)的制备方法
化合物1(R1=CH3)16.2g(0.10mo)l和盐酸羟胺10.3g(0.15mol)混合,溶于乙醇-水(v:v=1:1)180mL,加热至回流,加入氢氧化钠溶液[NaOH 6.8g(0.17mol)溶于50mL水],继续搅拌1h,TLC检测反应完全。停止加热,冰浴中冷却。抽滤,干燥得红色固体,无水乙醇重结晶,得到白色晶体15.7g,收率88.7%。
实施实例2:
化合物3(R1=CH3)的制备方法
化合物2(R1=CH3)3.54g(20mmol)溶于甲醇40mL,加NiCl2·6H2O 0.48g(0.02mmol),搅拌溶解,置冰浴中,N2保护下分批缓慢加入NaBH43.05g(80mmol),控制温度低于10℃,加毕继续搅拌2h,反应完全。加水100mL,用6N盐酸调整溶液至pH约为2,50℃搅拌2h,撤去氮气保护,减压蒸除甲醇,加氨水调至碱性,二氯甲烷萃取3次,饱和食盐水洗,无水硫酸钠干燥,蒸除溶剂,得到黄色油状液体2.54g,收率77.9%。
实施实例3:化合物4的制备方法1
氮气保护下,锥形瓶中加化合物3(R1为CH3)3.26g(0.02mol)、2-甲氧基乙酸异丙酯.9g(0.024mol)、无水碳酸钠1.0g和固定化CALB 1.0g,加干燥甲苯20mL,放入60℃摇床反应8h,过滤回收固定化CALB和Pd/AlO(OH),滤液蒸除溶剂,产物用石油醚/乙酸乙酯=2:1重结晶,得到白色固体,收率82.6%,熔点95.2-96.5℃,产品ee%>99%。
实施实例4:化合物4的制备方法2
氮气保护下,锥形瓶中加化合物3(R1为CH3)3.26g(0.02mol)、乙酸异丙烯酯2.4g(0.024mol)、无水碳酸钠1.0g和固定化CALB 1.0g,加干燥甲苯20mL,放入70℃摇床反应6h,过滤回收固定化CALB和Pd/AlO(OH),滤液蒸除溶剂,产物用石油醚/乙酸乙酯=2:1重结晶,得到淡黄色固体,收率56.4%,ee%为70%。
实施实例5:化合物4的制备方法3
氮气保护下,锥形瓶中加化合物3(R1为CH3)3.26g(0.02mol)、2-甲氧基乙酸异丙酯 2.9g(0.024mol)、无水碳酸钠1.0g和固定化CALB 1.0g,加丙酮20mL,放入60℃摇床反应24h,过滤回收固定化CALB和Pd/AlO(OH),滤液蒸除溶剂,产物用石油醚/乙酸乙酯=2:1重结晶,得到白色固体,收率38.2%,ee%为80%。
实施实例6:化合物4的制备方法4
氮气保护下,锥形瓶中加化合物3(R1为CH=CH2)3.42g(0.02mol)、2-甲氧基乙酸异丙酯2.9g(0.024mol)、无水碳酸钠1.0g和固定化CALB1.0g,加干燥甲苯20ml,放入60℃摇床、反应24h,过滤回收固定化CALB和Pd/AlO(OH),滤液蒸除溶剂,产物用石油醚/乙酸乙酯=2:1重结晶,得到白色固体,收率42.1%,ee%为83%。
实施实例7:化合物4的制备方法5
氮气保护下,锥形瓶中加化合物3(R1为CH3)3.26g(0.02mol)、2-甲氧基乙酸异丙酯2.9g(0.024mol)、无水碳酸钠1.0g和固定化CALB1.0g,加干燥甲苯20ml,放入60℃摇床反应24h,过滤回收固定化CALB和Pd/BaSO4,滤液蒸除溶剂,产物用石油醚/乙酸乙酯=2:1重结晶,得到白色固体,收率53%,ee%为68%。
实施实例8:化合物4的制备方法6
氮气保护下,锥形瓶中加化合物3(R1为CH3)3.26g(0.02mol)、2-甲氧基乙酸异丙酯2.9g(0.024mol)、无水碳酸钠1.0g和固定化CALA1.0g,加干燥甲苯20ml,放入60℃摇床反应24h,过滤回收固定化CALA和Pd/AlO(OH),滤液蒸除溶剂,产物用石油醚/乙酸乙酯=2:1重结晶,得到白色固体,收率25%,ee%为56%。
实施实例9:化合物4的制备方法7
氮气保护下,锥形瓶中加化合物3(R1为CH3)3.26g(0.02mol)、2-甲氧基乙酸异丙酯2.9g(0.024mol)、无水碳酸钠1.0g和固定化CALB1.0g,加干燥甲苯20ml,放入20℃摇床反应24h,过滤回收固定化CALB和Pd/AlO(OH),滤液蒸除溶剂,产物用石油醚/乙酸乙酯=2:1重结晶,得到白色固体,收率60%,ee%为75%。
- 还没有人留言评论。精彩留言会获得点赞!