一种抗痛风药非布司他的合成方法与流程
本发明涉及抗痛风药的合成,具体涉及一种抗痛风药非布司他的合成方法。
背景技术:
非布司他是一种抗痛风药,其作用机制是药物分子占据黄嘌呤氧化酶(XO)的疏水空腔,抑制了酶的催化活性,减少了嘌呤向尿酸的转化,从而达到治疗痛风的目的。此外,非布司他是一种非嘌呤类化合物,对XO具有较高的选择性,不会作用于嘌呤和嘧啶代谢途径中相关的酶而产生相应的核苷类代谢物,因此不会影响嘌呤与嘧啶的正常代谢,有较高的安全性,市场应用前景广阔。
非布司他(Febuxostat),化学名为2-(3-氰基-4-异丁氧基苯基)-4-甲基-5-噻唑甲酸,分子式为C16H16N2O3S,分子量为316.37,其化学结构是如下所示:
目前有关非布司他的文献报道较多,具有代表性的文献是:
(1)Kondo S,Fukushima H,Hasegawa M,et al.2-Arylthiazole derivative and pharma-ceutical composition containing the same:WO,9209279[P].1992-06-11(CA 1993,118:38915)
(2)漆又毛,揭清,张冯敏。抑制黄嘌呤氧化酶活性的芳腈基噻唑衍生物及制备方法和用途,中国.10138664[P].2009-03-18(CA2009,150:423167)。
(3)Minoshima T,Hiramatsu T.Preparation of 2-(3-cyanophenyl)thiazoles as pharmac-euticals for treatment of gout and hyperuricemia:JP,10139770[J].1998-05-26.(CA 1998,129:54365)。
以上是比较经典的合成目标化合物非布司他的路线,但都存在以下缺点:
(1)合成过程中使用剧毒物质KCN,对操作人员和环境构成严重威胁。
(2)起始原料和中间体用到的部分试剂价格较高,且收率较低,所以整条路线成本比较高。
整体来说,目前非布司他的合成方法存在环境污染严重,成本较高,不适合工业生产的缺点。
技术实现要素:
本发明目的在于提供一种抗痛风药非布司他的合成方法。
为实现上述目的,本发明采用技术方案为:
一种抗痛风药非布司他的合成方法,以对羟基苯甲酸乙酯为原料,通过Duff反应引入醛基,经Williamson醚化、氰化、水解氨化、硫化、环合、再经水解反应即得到化合物1非布司他;合成路线如下:
按照65-75wt%的多聚磷酸:对羟基苯甲酸乙酯的质量比为4-5:1的比例混合,搅拌过程中分批加入乌洛托品,升温到75-85℃,同温反应3-5h,待反应结束后,反应液依次用过量的水,乙酸乙酯反复萃取,收集的有机相用过量的饱和食盐水洗2-3次后有机相浓缩,固体40-60℃烘干得3-甲酰基-4-羟基苯甲酸乙酯(化合物2);其中,乌洛托品:对羟基苯甲酸乙酯的摩尔比为0.5-2.5:1。
所述多聚磷酸预先用水稀释,待体系降到20-25℃使用,以免反应剧烈放热发生危险。
搅拌条件下,将3-甲酰基-4-羟基苯甲酸乙酯溶解在DMF中,然后向体系中加入碳酸钾和碘化钾;避光下,将溴代异丁烷与DMF按照体积比1:1-2的比例混合得混合液,而后将混合液滴入到上述反应体系中,滴加完毕后70-85℃反应5-7h,反应结束后趁热过滤,滤液降温到-5-5℃析晶5-7h,析晶后过滤滤饼40-60℃烘干得3-甲酰基-4-异丁氧基苯甲酸乙酯(化合物3);其中,碳酸钾:3-甲酰基-4-羟基苯甲酸乙酯的摩尔比4-6:1;碘化钾:3-甲酰基-4-羟基苯甲酸乙酯的摩尔比4-6:1;溴代异丁烷:3-甲酰基-4-羟基苯甲酸乙酯的摩尔比1-5:1,DMF(溶解3-甲酰基-4-羟基苯甲酸乙酯的DMF):3- 甲酰基-4-羟基苯甲酸乙酯的体积质量比为6-10L:1Kg。
所述滴加速度为80-100mL/h。
在搅拌条件下,将3-甲酰基-4-异丁氧基苯甲酸乙酯:甲酸的质量比为1:8-11的比例混合均匀,向反应体系中加入盐酸羟胺和甲酸钠,升温到90-105℃,回流反应3-5h,反应完毕后,加入过量的水使反应体系降温到-5-5℃,有固体析出,过滤,滤饼40-60℃烘干得3-氰基-4-异丁氧基苯甲酸乙酯(化合物4);其中,盐酸羟胺:3-甲酰基-4-异丁氧基苯甲酸乙酯的摩尔比为1.1-1.5:1;甲酸钠:3-甲酰基-4-异丁氧基苯甲酸乙酯的摩尔比为1.1-1.5:1。
在搅拌条件下,将3-氰基-4-异丁氧基苯甲酸乙酯加入到过量的乙醇中,再加入氢氧化钠水溶液,升温到40-60℃,反应2-5h,反应完毕后减压蒸除乙醇,调节体系pH=5-7,析出固体,过滤得3-氰基-4-异丁氧基苯甲酸;将3-氰基-4-异丁氧基苯甲酸溶于过量的二氯甲烷中,加入DMF后滴加二氯亚砜,而后反应1-3h,反应后加入过量的THF,加入后降温到-5-5℃,再滴加过量的25-28%的氨水,同温反应1-3h后蒸除THF析出固体,过滤,滤饼40-60℃烘干得3-氰基-4-异丁氧基苯甲酰胺(化合物5);其中,乙醇:3-氰基-4-异丁氧基苯甲酸乙酯的体积质量比为5-10L:1Kg;氢氧化钠:3-氰基-4-异丁氧基苯甲酸乙酯的摩尔比为1.1-2.5:1;二氯甲烷:3-氰基-4-异丁氧基苯甲酸乙酯的体积质量比为8-10L:1Kg;二氯亚砜:3-氰基-4-异丁氧基苯甲酸乙酯的体积质量比为1-2L:1Kg;THF:3-氰基-4-异丁氧基苯甲酸乙酯的体积质量比为4-6.5L:1Kg;25-28%的氨水:3-氰基-4-异丁氧基苯甲酸乙酯的体积质量比为5-8L:1Kg。
在搅拌条件下,将3-氰基-4-异丁氧基苯甲酰胺加入到过量的THF中,加入Lawesson试剂,氮气保护下升温到60-70℃,反应5-8h,反应完毕,蒸馏蒸除约3/4THF,而后再于搅拌条件下依次经过量的NaHCO3饱和溶液,二氯甲烷反复萃取,合并萃取所得的有机相,有机相用过量的饱和食盐洗涤,洗涤后浓缩得3-氰基-4-异丁氧基硫代苯甲酰胺(化合物6);其中,Lawesson试剂:3-氰基-4-异丁氧基苯甲酰胺的摩尔比0.4-0.6:1。
将3-氰基-4-异丁氧基硫代苯甲酰胺加入到过量的乙醇中,在搅拌条件下加入2-氯乙酰乙酸乙酯,而后升温反应体系的温度到75-85℃,反应2-5h,反应完毕后蒸除约3/4乙醇,降温到-5-5℃,析出固体,过滤,滤饼40-60℃烘干得2-(3-氰基-4-异丁氧基苯基)-4-甲基-5-噻唑甲酸乙酯(化合物7);其中,2-氯乙酰乙酸乙:3-氰基-4-异丁氧基硫代苯甲酰胺的摩尔比1.1-1.5:1。
搅拌条件下将2-(3-氰基-4-异丁氧基苯基)-4-甲基-5-噻唑甲酸乙酯溶解到乙醇与THF中的混合溶剂中,溶解后滴加NaOH水溶液, 滴加完毕升温到40-60℃,反应2-4h,而后调节反应液pH=5,使体系中析出固体,过滤,滤饼用过量的95%乙醇重结晶,得2-(3-氰基-4-异丁氧基苯基)-4-甲基-5-噻唑甲酸(化合物1);其中,乙醇:2-(3-氰基-4-异丁氧基苯基)-4-甲基-5-噻唑甲酸乙酯的体积质量比为3-5L:1Kg;THF:2-(3-氰基-4-异丁氧基苯基)-4-甲基-5-噻唑甲酸乙酯的体积质量比为2-4L:1Kg;NaOH:2-(3-氰基-4-异丁氧基苯基)-4-甲基-5-噻唑甲酸乙酯的摩尔比为1.1-1.5:1。
与现有的合成技术相比,本发明具有如下优点:
(1)本发明合成方法中将Duff引入醛基,现有技术使用CF3COOH作为酸性介质,本发明用多聚磷酸为溶剂,不仅提高收率,避免使用剧毒物质,也简化后处理方式,同时降低成本,适合工业生产。
(2)本发明合成方法中合成化合物5时,现有技术氨化不彻底,还有部分原料剩余,氨水的量增大后,反应更加彻底,且产物不用提纯即可进行后续反应。
(3)本发明合成各步反应溶剂使用了环境友好的溶剂,同时都可回收利用,节约成本,减少对环境的破坏。
(4)本发明以对羟基苯甲酸乙酯为原料,经Duff反应、醚化、氰化、水解氨化、硫化、环合、水解7步反应,合成非布司他,各步收率都比较高,总收率为25-30%。
附图说明
图1为本发明实施例提供的3-甲酰基-4-羟基苯甲酸乙酯的1H NMR图。
图2为本发明实施例提供的3-甲酰基-4-异丁氧基苯甲酸乙酯的 1H NMR图。
图3为本发明实施例提供的3-氰基-4-异丁氧基苯甲酸乙酯的1H NMR图。
图4为本发明实施例提供的3-氰基-4-异丁氧基苯甲酰胺的1H NMR图。
图5为本发明实施例提供的2-(3-氰基-4-异丁氧基苯基)-4-甲基-5-噻唑甲酸乙酯的1H NMR图。
图6为本发明实施例提供的2-(3-氰基-4-异丁氧基苯基)-4-甲基-5-噻唑甲酸的1H NMR图。
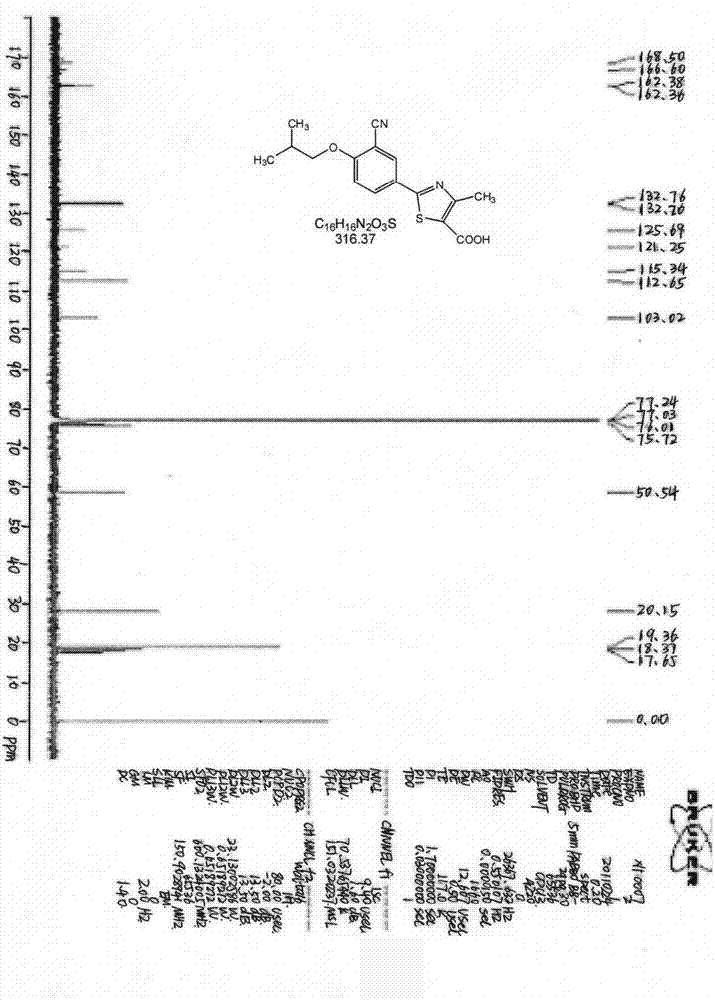
图7为本发明实施例提供的2-(3-氰基-4-异丁氧基苯基)-4-甲基-5-噻唑甲酸的13C NMR图。
图8为本发明实施例提供的2-(3-氰基-4-异丁氧基苯基)-4-甲基-5-噻唑甲酸的MS图。
具体实施方式
通过以下实施例具体实施,对本发明的上述内容作进一步的说 明。
实施例1
(1)制备3-甲酰基-4-羟基苯甲酸乙酯:
在装有电动搅拌器、干燥管(内装无水氯化钙)和温度计的500mL三颈烧瓶中,加入33.60g(0.20mol)对羟基苯甲酸乙酯,56.80g(0.41mol)乌洛托品和150.00g 75wt%的多聚磷酸,于82℃下搅拌反应3-5h,反应停止后,加水80mL,乙酸乙酯(3×80mL)萃取,有机相用饱和食盐水(2×60mL)洗涤,有机相浓缩,得白色固体26.78g化合物2(参见图1),收率68.20%,m.p.67-68℃。 1H NMR(CDCl3),δ:1.41(t,3H,J=7.2Hz);4.38-4.41(q,2H);7.04(d,1H,J=9.0Hz);8.20(dd,1H,J=2.4Hz,J=9.0Hz);8.33(d,1H,J=2.4Hz);9.96,(s,1H);11.39(s,1H)。
(2)制备3-甲酰基-4-异丁氧基苯甲酸乙酯:
在装有恒压滴液漏斗、冷凝管和温度计的500mL三颈烧瓶中,加入15.30g(78.94mmol)化合物2,54.60g(0.39mol)碳酸钾,6.55g(0.39mol)碘化钾和150mLDMF,搅拌下升温至72℃,缓慢滴加45mL(0.39mol)溴代异丁烷和65mLDMF的混合溶液,滴毕,同温(72℃)反应6h,停止反应后,趁热过滤,滤液降温-5-5℃析晶,得白色固体15.61g化合物3(参见图2),收率79.00%,m.p.66.5-68.5℃。1H NMR(CDCl3),δ:1.09(d,6H,J=6.6Hz);1.39(t,3H,J=7.2Hz);2.19-2.21(m,1H);3.92(d,2H,J=6.6Hz);4.35-4.39(q,2H);7.02(d,1H,J=9.0Hz);8.23(dd,1H,J=2.4Hz,J=9.0Hz);8.51(d,1H,J=2.4Hz);10.53(s,1H)。
(3)制备3-氰基-4-异丁氧基苯甲酸乙酯:
在装有磁搅拌器、冷凝管和温度计的250mL三颈烧瓶中,加入170.00g甲酸,17.80g(0.07mol)化合物3,6.90g(0.10mol)盐酸羟胺和7.40g(0.10mol)甲酸钠,升温至102℃,回流反应约5h(TLC监测至原料点消失),停止反应,冷却至0-5℃,搅拌2h, 有固体析出,抽滤,滤饼用冰水淋洗,得白色固体14.01g化合物4(参见图3),收率79.78%,m.p.100.9-103.9℃。1H NMR(CDCl3),δ:1.09(d,6H,J=6.6Hz);1.40(t,3H,J=7.2Hz);2.18-2.23(m,1H);3.90(d,2H,J=6.6Hz);4.36-4.39(q,2H);6.98(d,1H,J=9.0Hz);8.12(dd,1H,J=2.4Hz,J=9.0Hz);8.26(d,1H,J=2.4Hz)。
(4)制备3-氰基-4-异丁氧基苯甲酰胺:
在装有磁搅拌器、冷凝管和温度计的250mL三颈烧瓶中,加入14.95g(0.06mol)化合物4,搅拌下分别加入85mL乙醇和60mL 2mol L-1的氢氧化钠溶液,升温至60℃,恒温反应3h,旋蒸除去乙醇,用1mol L-1盐酸调pH=5,析出固体,过滤,滤饼用少量冰水淋洗,干燥得白色固体11.83g。
将上述白色固体溶于120mL二氯甲烷中,加入DMF 0.5mL,搅拌下缓慢滴加12mL氯化亚砜,室温反应4h,旋蒸除去溶剂后残余物溶于65mL THF中,冰水浴冷却至0-5℃,滴加25%的氨水180mL,搅拌反应1.5h,浓缩得白色固体10.83g化合物5(参见图4),收率82.19%,m.p.124.6-127.6℃。1H NMR(CDCl3),δ:1.09(d,6H,J=6.6Hz);2.18-2.23(m,1H);3.90(d,2H,J=6.6Hz);5.69-6.00(br,s,2H);7.01(d,1H,J=9Hz);8.02-8.03(m,2H)。
(5)制备3-氰基-4-异丁氧基硫代苯甲酰胺:
在装有磁搅拌器、冷凝管(上口接无水氯化钙干燥管)和温度计的250mL三颈烧瓶中,加入10.80g(0.05mol)化合物5,11.00g(0.027mol)Lawesson试剂和110mL无水THF,氮气保护下,慢慢升温至回流,恒温反应6h(TLC监测至原料点消失),停止反应,蒸除溶剂后,加入40mL碳酸氢钠饱和溶液,二氯甲烷(3×40mL)萃取,有机相用饱和食盐水(2×30mL)洗涤,抽滤,滤液浓缩得化合物6(参见图5),直接用于下步反应。
(6)制备2-(3-氰基-4-异丁氧基苯基)-4-甲基-5-噻唑甲酸乙酯:
在装有磁搅拌器、冷凝管和温度计的100mL三颈烧瓶中,9.60g(0.04mol)化合物6溶于50mL乙醇中,搅拌条件下加入8.00g(0.048mol)2-氯乙酰乙酸乙酯,缓慢升温至回流,反应3h,旋蒸除去大部分乙醇,冰水浴冷却至0-5℃,搅拌1h,有固体析出,抽滤,滤饼冰乙醇淋洗,得白色固体12.40g化合物7(参见图6),收率88.12%,m.p.174-176℃。1H NMR(CDCl3),δ:1.09(d,6H,J=6.6Hz);1.39(t,3H,J=7.2Hz);2.20-2.22(m,1H);2.78(s,3H);3.90(d,2H,J=6.6Hz);4.34-4.38(q,2H);7.02(d,1H,J=9.0Hz);8.12(dd,1H,J=2.4Hz,J=9.0Hz);8.18(d,1H,J=2.4Hz)。
(7)制备2-(3-氰基-4-异丁氧基苯基)-4-甲基-5-噻唑甲酸:
在装有磁搅拌器、冷凝管和温度计的250mL三颈烧瓶中,加入11.95g(34.72mmol)化合物7,1.70g(41.75mmol)氢氧化钠和25mL水,再加入60mL乙醇和35mL THF的混合溶剂,搅拌下升温至55℃,恒温反应2h,停止加热,旋蒸除去溶剂,稀盐酸调节pH=5,有大量固体析出,抽滤,滤饼用少量冰乙醇淋洗,得粗品,100mL乙醇重结晶得纯品9.67g,收率88.06%,m.p(参见图7和图8).201-204℃。1H NMR(CDCl3,600MHz),δ:1.10(d,6H,J=6.6Hz);2.19-2.23(m,1H);2.81(s,3H);3.91(d,2H,J=6.6Hz);7.03(d,1H,J=9.0Hz);8.13(dd,1H,J=2.4Hz,J=9.0Hz);8.21(d 1H,J=2.4Hz);13.39(s,1H)。13C NMR(DCCl3,150MHz),δ:168.50(CN),166.60(COOH),162.78(C2),162.70(C4′),132.76(C6′),132.26(C2′),125.69(C3′),121.25(C1′),115.34(C5),112.65(C5′),103.02(C4),75.72[(CH3)2CH-CH2O],28.15[(CH3)2CH],19.06[(CH3)2CH],17.65(CH3)。ESI-MS(m/z): (314.81,M-)。
实施例2
(1)制备3-甲酰基-4-羟基苯甲酸乙酯:
在装有电动搅拌器、和温度计的500L反应釜中,加入50.00Kg(300.00mol)对羟基苯甲酸乙酯,56.80Kg(620.00mol)乌洛托品和220.00Kg 75wt%的多聚磷酸,于84℃下搅拌反应3h,反应停止后,加水120L,乙酸乙酯(3×120L)萃取,有机相用饱和食盐水(2×90mL)洗涤,有机相浓缩,得白色固体40.12Kg,收率68.87%。
(2)制备3-甲酰基-4-异丁氧基苯甲酸乙酯:
在装有恒压滴液漏斗、冷凝管和温度计的500L反应釜中,加入30.00Kg(154.47mol)化合物2,106.80Kg(760.00mol)碳酸钾,12.81Kg(760.00mol)碘化钾和290L DMF,搅拌下升温至74℃,缓慢滴加85L(390.00mol)溴代异丁烷和120L DMF的混合溶液,滴毕,同温反应6h,停止反应后,趁热压滤,滤液降温0-5℃析晶,得白色固体29.45Kg,收率76.17%。
(3)制备3-氰基-4-异丁氧基苯甲酸乙酯:
在装有磁搅拌器、冷凝管和温度计的500L反应釜中,加入235.00Kg甲酸,25.00Kg(100.00mol)化合物3,9.65Kg(140.00mol)盐酸羟胺和10.40g(140.00mol)甲酸钠,升温至100℃,回流反应约5h,停止反应,冷却至0-5℃,搅拌4h,有固体析出,离心,滤饼用水洗,得白色固体20.22Kg,收率81.86%。
(4)制备3-氰基-4-异丁氧基苯甲酰胺:
在装有磁搅拌器、冷凝管和温度计的500L反应釜中,加入20.00Kg(80.00mol)化合物4,搅拌下分别加入115L乙醇和80L 2molL-1的氢氧化钠溶液,升温至62℃,恒温反应3h,浓缩除去乙醇,用1mol L-1盐酸调pH=5,析出固体,离心,滤饼用水洗,干燥得白色固体16.23Kg。
将上述白色固体溶于160L二氯甲烷中,加入DMF 0.5L,搅拌下缓慢滴加16L氯化亚砜,20-25℃反应4h,旋蒸除去溶剂后残余物溶于85L THF中,冰水浴冷却至0-5℃,滴加25%的氨水240L,搅拌反应3h,浓缩得白色固体15.12Kg,收率86.60%。
(5)制备3-氰基-4-异丁氧基硫代苯甲酰胺:
在装有磁搅拌器、冷凝管和温度计的250L反应釜中,加入15.00Kg(70.00mol)化合物5,15.30Kg(37.00mol)Lawesson试剂和150L无水THF,氮气保护下,慢慢升温至回流,恒温反应6h,停止反应,蒸除溶剂后,加入55L碳酸氢钠饱和溶液,二氯甲烷(3×55L)萃取,有机相用饱和食盐水(2×40L)洗涤,抽滤,滤液浓缩,直接用于下步反应。
(6)制备2-(3-氰基-4-异丁氧基苯基)-4-甲基-5-噻唑甲酸乙酯:
在装有磁搅拌器、冷凝管和温度计的250L反应釜中,13.50Kg(50.00mol)化合物6溶于70L乙醇中,搅拌条件下加入11.50Kg(60.00mol)2-氯乙酰乙酸乙酯,缓慢升温至回流,反应4h,浓缩 除去大部分乙醇,冷却至0-5℃,搅拌1h,有固体析出,离心,滤饼乙醇洗,得白色固体17.90Kg,收率90.22%。
(7)制备2-(3-氰基-4-异丁氧基苯基)-4-甲基-5-噻唑甲酸:
在装有磁搅拌器、冷凝管和温度计的250L反应釜中,加入16.65Kg(48.27mol)化合物7,2.30Kg(58.00mol)氢氧化钠和35L水,再加入80L乙醇和50L THF的混合溶剂,搅拌下升温至52℃,恒温反应3h,停止加热,浓缩除去溶剂,稀盐酸调节pH=5,有大量固体析出,离心,滤饼用少量乙醇洗,得粗品,100L乙醇重结晶得纯品13.55Kg,收率88.62%。
实施例3
(1)制备3-甲酰基-4-羟基苯甲酸乙酯:
在装有电动搅拌器和温度计的1000L反应釜中,加入65.00Kg(390.00mol)对羟基苯甲酸乙酯,73.80Kg(810.00mol)乌洛托品和280.00Kg 75wt%的多聚磷酸,于80℃下搅拌反应3h,反应停止后,加150L水,乙酸乙酯(3×150L)萃取,有机相用饱和食盐水(2×120L)洗涤,有机相浓缩,得白色固体50.23Kg,收率66.33%。
(2)制备3-甲酰基-4-异丁氧基苯甲酸乙酯:
在装有恒压滴液漏斗、冷凝管和温度计的1000L反应釜中,加入40.00Kg(206.00mol)化合物2,138.80Kg(980.00mol)碳酸钾,16.65Kg(980.00mol)碘化钾和375LDMF,搅拌下升温至76℃, 缓慢滴加110L(510.00mol)溴代异丁烷和155LDMF的混合溶液,滴毕,同温反应5h,停止反应后,趁热过滤,滤液降温0-5℃析晶,得白色固体41.32Kg,收率80.13%。
(3)制备3-氰基-4-异丁氧基苯甲酸乙酯:
在装有磁搅拌器、冷凝管和温度计的1000L的反应釜中,加入305.00Kg甲酸,32.50Kg(130.00mol)化合物3,12.55Kg(180.00mol)盐酸羟胺和13.55Kg(180.00mol)甲酸钠,升温至103℃,回流反应约6h,停止反应,冷却至0-5℃,搅拌2h,有固体析出,离心,滤饼用水洗,得白色固体28.56Kg,收率88.94%。
(4)制备3-氰基-4-异丁氧基苯甲酰胺:
在装有磁搅拌器、冷凝管和温度计的500L反应釜中,加入25.00Kg(100.00mol)化合物4,搅拌下分别加入150L乙醇和105L 2mol L-1的氢氧化钠溶液,升温至65℃,恒温反应4h,浓缩除去乙醇,用1mol L-1盐酸调pH=5,析出固体,离心,滤饼用少量水洗,干燥得白色固体21.52Kg。
将上述白色固体溶于210L二氯甲烷中,加入DMF 0.5L,搅拌下缓慢滴加21L氯化亚砜,20-25℃反应5h,浓缩除去溶剂后残余物溶于110L THF中,冷却至-5-5℃,滴加25%的氨水310L,搅拌反应3h,浓缩得白色固体20.13Kg,收率91.61%。
(5)制备3-氰基-4-异丁氧基硫代苯甲酰胺:
在装有磁搅拌器、冷凝管和温度计的500L反应釜中,加入20.00Kg(90.00mol)化合物5,20.00Kg(49.00mol)Lawesson试剂和 200L无水THF,氮气保护下,慢慢升温至回流,恒温反应8h,停止反应,浓缩溶剂后,加入75L碳酸氢钠饱和溶液,二氯甲烷(3×75L)萃取,有机相用饱和食盐水(2×55L)洗涤,抽滤,滤液浓缩,直接用于下步反应。
(6)制备2-(3-氰基-4-异丁氧基苯基)-4-甲基-5-噻唑甲酸乙酯:
在装有磁搅拌器、冷凝管和温度计的250L反应釜中,15.00Kg(55.00mol)化合物6溶于75L乙醇中,搅拌条件下加入12.75Kg(60.00mol)2-氯乙酰乙酸乙酯,缓慢升温至回流,反应3h,浓缩除去大部分乙醇,冷却至0-5℃,搅拌2h,有固体析出,离心,滤饼用乙醇洗,得白色固体18.20Kg,收率91.70%。
(7)制备2-(3-氰基-4-异丁氧基苯基)-4-甲基-5-噻唑甲酸:
在装有磁搅拌器、冷凝管和温度计的250L反应釜中,加入17.50Kg(50.68mol)化合物7,2.41Kg(60.90mol)氢氧化钠和35L水,再加入85L乙醇和55L THF的混合溶剂,搅拌下升温至55℃,恒温反应2h,停止加热,浓缩除去溶剂,稀盐酸调节pH=5,有固体析出,离心,滤饼用少量乙醇淋洗,得粗品,100L乙醇重结晶得纯品14.08Kg,收率87.83%。
实施例4
(1)制备3-甲酰基-4-羟基苯甲酸乙酯:
在装有电动搅拌器和温度计的1000L反应釜中,加入80.00Kg(480.00mol)对羟基苯甲酸乙酯,90.70Kg(990.00mol)乌洛托品和345.00Kg 75wt%的多聚磷酸,于80℃下搅拌反应5h,反应停止后,加185L水,乙酸乙酯(3×180L)萃取,有机相用饱和食盐水(2×150L)洗涤,有机相浓缩,得白色固体62.32Kg,收率67.00%。
(2)制备3-甲酰基-4-异丁氧基苯甲酸乙酯:
在装有恒压滴液漏斗、冷凝管和温度计的2000L反应釜中,加入49.20Kg(253.38mol)化合物2,170.70Kg(1200mol)碳酸钾,20.50Kg(1200mol)碘化钾和450L DMF,搅拌下升温至75℃,缓慢滴加135L(510.00mol)溴代异丁烷和190L DMF的混合溶液,滴毕,同温反应6h,停止反应后,趁热过滤,滤液降温-5-5℃析晶,得白色固体51.56Kg,收率81.31%。
(3)制备3-氰基-4-异丁氧基苯甲酸乙酯:
在装有磁搅拌器、冷凝管和温度计的1000L反应釜中,加入375.00Kg甲酸,40.00Kg(160.00mol)化合物3,15.40Kg(220.00mol)盐酸羟胺和16.67Kg(220.00mol)甲酸钠,升温至104℃,回流反应约5h,停止反应,冷却至0-5℃,搅拌4h,有固体析出,离心,滤饼用冰水淋洗,得白色固体34.37Kg,收率87.13%。
(4)制备3-氰基-4-异丁氧基苯甲酰胺:
在装有磁搅拌器、冷凝管和温度计的500L反应釜中,加入30.75Kg(120.00mol)化合物4,搅拌下分别加入185L乙醇和130L 2mol L-1的氢氧化钠溶液,升温至61℃,恒温反应3h,旋蒸除去乙醇, 用1mol L-1盐酸调pH=5,析出固体,离心,滤饼用少量冰水淋洗,干燥得白色固体26.12Kg。
将上述白色固体溶于260L二氯甲烷中,加入DMF 0.5L,搅拌下缓慢滴加26L氯化亚砜,20-25℃反应4h,浓缩除去溶剂后残余物溶于135L THF中,冷却至0-5℃,滴加25%的氨水380L,搅拌反应1.5h,浓缩得白色固体23.45Kg,收率89.53%。
(5)制备3-氰基-4-异丁氧基硫代苯甲酰胺:
在装有磁搅拌器、冷凝管和温度计的500L反应釜中,加入25.00Kg(110.00mol)化合物5,25.00Kg(60.00mol)Lawesson试剂和250L无水THF,氮气保护下,慢慢升温至回流,恒温反应6h,停止反应,蒸除溶剂后,加入95L碳酸氢钠饱和溶液,二氯甲烷(3×95L)萃取,有机相用饱和食盐水(2×65L)洗涤,压滤,滤液浓缩,直接用于下步反应。
(6)制备2-(3-氰基-4-异丁氧基苯基)-4-甲基-5-噻唑甲酸乙酯:
在装有磁搅拌器、冷凝管和温度计的250L反应釜中,18.50Kg(60.00mol)化合物6溶于95L乙醇中,搅拌条件下加入15.65Kg(70.00mol)2-氯乙酰乙酸乙酯,缓慢升温至回流,反应5h,浓缩除去大部分乙醇,冷却至0-5℃,搅拌3h,有固体析出,离心,滤饼用乙醇洗,得白色固体23.45Kg,收率86.24%。
(7)制备2-(3-氰基-4-异丁氧基苯基)-4-甲基-5-噻唑甲酸:
在装有磁搅拌器、冷凝管和温度计的250L反应釜中,加入21.50Kg(62.33mol)化合物7,2.95Kg(73.75mol)氢氧化钠和45L水,再加入105L乙醇和65L THF的混合溶剂,搅拌下升温至50℃,恒温反应2h,停止加热,旋蒸除去溶剂,稀盐酸调节pH=5,有大量固体析出,离心,滤饼用少量乙醇洗,得粗品,125L乙醇重结晶得纯品17.51Kg,收率88.66%。
最后说明的是尽管参照较佳的实例对本方案进行详尽的说明,但不局限于此,只是用于帮助理解本发明,对于本技术领域的人员而言,在不脱离本发明原理的基础上,还可以对本发明进行若干改进,而这些改进也落入本发明权利要求的保护范围内。
- 还没有人留言评论。精彩留言会获得点赞!