用于确定患有BRAF‑阳性癌症的患者为BRAF抑制剂非响应者且为MAPK/ERK抑制剂响应者的工具和方法与流程
本发明涉及诊断学领域,特别是癌症诊断。更具体地,本发明涉及用于确定患有braf-阳性癌症的对象是否为braf抑制剂的非响应者,和/或mapk/erk抑制剂的响应者,用于诊断癌症的方法,用于评估对象对靶向疗法的响应性的方法,以及用于评估对象的癌症的方法。此外,本发明还涉及用于诊断癌症的试剂盒和设备。进一步地,本发明涉及mapk/erk抑制剂,其用于治疗患有braf-阳性癌症的对象。
用于晚期疾病的黑素瘤疗法在最近几年取得了巨大的进展1-3,但一些患者对靶向疗法原生内在抗性,以及在大多数其他患者中的延迟获得性抗性的发作,仍构成转移性黑素瘤的临床管理的主要挑战4。
然而,下一代测序(ngs)技术的出现允许解决常规疗法如何影响患者体内遗传变异的异质情形,以及确定治疗抗性的来源的问题。除了阐明癌症进展的新机制,ngs应用还提供了用于随时间的克隆多样性变化的量化和建模的大型数据集。在一些癌症中,整体遗传多样性量度已显示预示肿瘤进展5。
特别地,转移性黑素瘤具有任何癌症中最高的突变率之一6。一些研究已经确定基因组特征,例如在原位瘤和转移瘤之间不同的异质性损失,其他研究已显示这种遗传异质性也存在于单个肿瘤内。
在治疗抗性背景下,现已在大量患者群之间证实了对靶向治疗响应的很多遗传和转录机制,但对系统治疗的个体癌症基因组进化仍了解不足9,10。已显示少数亚克隆展示出对疗法的敏感性降低7,并且更近期的研究已揭示接受靶向braf抑制剂的患者具有不同的抗性机制,其源自这种潜在的肿瘤内分子异质性。
通常可以区分两种不同的治疗抗性机制:内在(原生)的和获得(次生)的。内在抗性肿瘤或者一开始就不响应或者包含抗性亚克隆,其在治疗期间被快速地选择,导致不能减小肿瘤负荷和快速复发。获得性抗性机制在治疗期间产生,并且可包括mapk途径的基因中的其他活化突变的选择和发生10,24,25或mapk抑制剂中的失活突变26。另外,还报道了braf转录物的可变剪接和其他非遗传机制在治疗抗性中发挥作用27。尽管有大量研究涉及这个问题,但已知抗性机制的列表远远没有完成,并且在很多个体病例中,抗性的机制还是未知的。
活化braf或nras突变常见于人黑素瘤中。尽管braf和nras活化突变可共存于相同的黑素瘤中,但认为在单细胞水平上它们是相互排斥的45。此外,nras突变或braf突变的存在与明显的体外和体内生长性质相关,并可直接影响突变黑素瘤的临床管理45。
根据上述肿瘤抗性机制,非常期望表征用于适合的治疗干预的癌症,并且特别是关于其响应braf抑制剂治疗的能力。
可见,本发明的主要技术问题是提供遵照上述需求的工具和方法。所述技术问题通过权利要求书和下文中表征的实施方式解决。
因此,本发明涉及用于确定患有braf-阳性癌症的对象是否为braf抑制剂非响应者和/或为mapk/erk抑制剂响应者的方法,所述方法包括以下步骤:
(a)测定所述对象的样品中是否至少在nras基因中存在至少一个突变;和
(b)如果已测定出在nras基因中的所述至少一个突变,则将所述对象确定为braf抑制剂非响应者和mapk/erk抑制剂响应者。
本发明的方法优选为离体方法。此外,其可包括上文明确提及的那些之外的步骤。例如,进一步的步骤可涉及样品预处理或对所述方法获得的结果的评价。所述方法可以手工或由自动操作辅助进行。优选地,步骤(a)和/或(b)可以完全或部分地由自动操作辅助,例如,通过用于步骤(a)的测定的适合的机器人和传感设备,和/或用于步骤(b)中的确定的在数据处理设备上的计算机实现的计算算法。
本文使用的术语“确定”是指评估所述对象是否为braf抑制剂的非响应者,或是否为braf抑制剂的响应者。因此,确定的目标可以在于将对象归入非响应者的组,或将其从所述组排除。同样,确定的目标可以在于将对象归入响应者的组,或将其从所述组排除。此外,确定还涵盖所述对象是mapk/erk抑制剂响应者的评估。如本领域中那些技术人员所理解的,此类评估通常并非意在对于100%的待研究对象都是准确的。然而,此术语需要所述评估对某部分的对象(例如,群组研究中的群组)是准确的。本领域技术人员可使用各种熟知的统计评估工具,例如置信区间的确定、p值测定、student'st检验、mann-whitney检验等,测定一部分是否在统计学上显著,而无需再费周折。细节见于dowdy和wearden,statisticsforresearch,johnwiley&sons,newyork1983。优选的置信区间是至少90%、至少95%、至少97%、至少98%或至少99%。p值优选为0.1、0.05、0.01、0.005,或0.0001。
本文使用的术语“对象”是指动物,通常为哺乳动物,且更通常为人。根据本发明,所述对象应患有braf-阳性癌症。
如本文使用的“braf-阳性癌症”是指癌症,其包含通常源自单细胞克隆,具有braf活性的损害的癌细胞。
典型地,braf活性升高,导致所述细胞中的尤其是mapk-途径的活化。更典型地,braf活化是由braf基因中的至少一个突变导致的,其导致例如组成型活性的braf蛋白,或细胞内不再能受到控制的braf蛋白。导致活化的braf蛋白的具体braf突变在本文他处说明。一方面,所述对象可接受或未接受braf抑制剂治疗。根据本发明,典型的braf-阳性癌症是黑素瘤癌、非霍奇金淋巴瘤癌、结肠直肠癌、乳头状甲状腺癌、非小细胞肺癌、毛细胞白血病或肺腺癌。更典型地,其为黑素瘤癌。
术语“braf抑制剂”是指能够干扰braf活性的分子。braf抑制剂可以是特异性结合braf蛋白并且抑制其活性的抗braf抗体。此外,braf抑制剂可以是抑制性核酸。抑制性核酸可以是特异性结合braf蛋白并抑制其活性的适配体。其他抑制性核酸可以结合braf转录物,并抑制其翻译或使其降解。典型地,此类抑制性核酸可以是反义核酸、吗啉代寡核苷酸、抑制性rna分子,例如sirna或微小rna或核酶。
反义核酸分子通常为rna,并且包含与目标转录物基本或完美互补的核酸序列。一方面,反义核酸分子基本由核酸序列组成,所述核酸序列与目标转录物的至少100个连续核苷酸,更优选至少200、至少300、至少400或至少500个连续核苷酸互补。如何生成和使用反义核酸分子是本领域中熟知的(参见例如weiss,b.(ed.):antisenseoligodeoxynucleotidesandantisenserna:novelpharmacologicalandtherapeuticagents,crcpress,bocaraton,fl,1997)。吗啉代寡核苷酸是合成的核酸分子,其具有20至30个核苷酸,且通常为25个核苷酸的长度。
吗啉代类(morpholinos)通过标准的核酸碱基配对结合目标转录物的互补序列。它们具有标准的核酸碱基,其结合至吗啉环而不是脱氧核糖环,并且通过磷酸二酰胺基团而不是磷酸连接(参见,例如,summerton1997,antisense&nucleicaciddrugdevelopment7*(3):187-95)。由于使用不带电的磷酸二酰胺基团代替带负电荷的磷酸,消除了在通常的生理ph值范围内的离子化,所以吗啉代类在生物或细胞内为不带电的分子。吗啉代类的完整骨架由这些修饰的亚基制成。与抑制性小rna分子不同,吗啉代类不将其目标rna分子降解。相反,它们在空间上阻断与rna内目标序列的结合,并简单地阻碍本可以与所述rna相互作用的分子(参见例如summerton1999,biochimicaetbiophysicaacta1489(1):141–58)。
小干扰rna(sirnas)与编码目的基因的靶rna互补,并且通过rna干扰(rnai)减少或消除基因表达。类似地,微小rna包含互补的rna靶向序列,并且也通过rnai机制发挥作用。不受理论束缚,rnai通常用于通过靶向mrna沉默基因表达。简言之,细胞中的rnai过程由双链rna(dsrna)启动,dsrna被核糖核酸酶切割,从而产生sirna双链体。sirna结合另一细胞内酶复合体,由此将其活化以靶向与所述sirna序列同源(或互补)的任何mrna分子。所述复合体的功能是通过sirna链之一与靶mrna之间的碱基配对靶向所述同源mrna分子。随后,从所述sirna的3'末端约12个核苷酸处切割mrna并降解。以这种方式,可靶向并降解特异性mrna,从而导致来自靶向mrna的蛋白表达丢失。如本文使用的互补核苷酸序列是指rna链上的区域,其与靶基因的一部分的rna转录物互补。dsrna是指具有双链体结构的rna,其包含两条互补且反平行的核酸链。未必dsrna的所有的核苷酸都展示完全的watson-crick碱基配对;所述两条rna链可以是基本互补的。形成dsrna的rna链可以具有相同或不同数量的核苷酸,并且碱基配对的最大数量为dsrna的最短链中的核苷酸数量。优选地,所述dsrna的长度不超过49个,更优选少于25个,且最优选长为19-23个核苷酸。这个长度的dsrna在使用rnai技术抑制靶基因表达中特别有效。dsrna随后被核糖核酸酶降解成小干扰rna(sirna)。所述sirna的互补区允许sirna与靶rna的充分杂交,并且由此介导rnai。在哺乳动物细胞中,sirna长为约21-25个核苷酸。sirna序列需要足够长,以通过互补碱基配对相互作用使所述sirna与靶rna聚集在一起。与本发明的tet表达系统一起使用的sirna可以具有不同的长度。所述sirna的长度优选大于或等于10个核苷酸,并且足够长以与靶rna稳定相互作用;具体为15-30个核苷酸;更具体为15至30个核苷酸之间的任何整数,最优选15、16、17、18、19、20、21、22、23、24、25、26、27、28、29和30。所述足够长,是指大于或等于15个核苷酸的寡核苷酸,其具有足以在预期条件下提供期望功能的长度。所述稳定相互作用,是指小干扰rna与靶核酸的相互作用(例如,通过在生理条件下与靶标中的互补核苷酸形成氢键)。通常,这种互补性在sirna和rna靶标之间是100%,但如果期望,可以更低,优选91%、92%、93%、94%、95%、96%、97%、98%或99%。例如,21个碱基中的19个碱基可以是碱基配对的。在期望在各种等位基因变体之间进行选择的一些情况下,需要与靶基因100%互补,以有效辨别靶序列与其他等位基因序列。在等位基因靶标之间进行选择的情况下,长度的选择也是重要因素,因为其为涉及互补百分比以及区分等位基因之间差别的能力的其他因素。涉及使用rnai以沉默生物体(包括秀丽隐杆线虫(c.elegans)、果蝇、植物和哺乳动物)中的基因的方法是本领域中已知的(参见例如fire1998,nature391:806-811;fire1999,trendsgenet.15,358-363;sharp2001,genesdev.15,485-490;hammond2001,naturerev.genet.2,1110-1119;tuschl2001,chem.biochem.2,239-245;hamilton1999,science286,950-952;hammond2000,nature404,293-296;zamore2000,cell101,25-33;bernstein2001,nature409,363-366;elbashir2001,genesdev.15,188-200;wo0129058;wo09932619;和elbashir2001,nature411:494-498)。.
核酶是具有良好定义的三级结构的催化性rna分子,其允许催化其自身磷酸二酯键之一的水解(自裂解核酶),或其他rna中的键的水解,但也发现它们催化核糖体的氨基转移酶活性。根据本发明预想的核酶优选为特异性水解靶转录物的那些。具体地,根据本发明优选锤头核酶(hammerheadribozymes)。如何产生及使用此类核酶是本领域中熟知的(参见例如heanj,weinbergms(2008)"thehammerheadribozymerevisited:newbiologicalinsightsforthedevelopmentoftherapeuticagentsandforreversegenomicsapplications".inmorriskl.rnaandtheregulationofgeneexpression:ahiddenlayerofcomplexity.norfolk,england:caisteracademicpress)。
此外,braf抑制剂可以是结合braf并抑制其活性的小分子。braf的此类小分子抑制剂可以通过熟知的筛选程序或分子建模方法获得,所述分子建模方法的目的是确定与braf激酶结构域的活性位点结合的化合物。bay43-9006(也被称作索拉非尼(sorafenib)或多吉美(nexavar))是通过结合非活性形式的激酶结构域并阻断其活化而抑制braf活性的小分子。plx4032(也被称作维罗非尼(vemurafenib))是braf抑制剂,其将其本身锚定在激酶结构域的atp结合口袋中,并且由此阻断活性酶的活性。一方面,本文所述的braf抑制剂选自以下:lgx818(恩可非尼(encorafenib))、plx4032(维罗非尼)、gsk2118436(达帕非尼(dabrafenib))、gdc-0879,和bay43-9006(索拉非尼)。更典型地,所述braf抑制剂是lgx818(恩可非尼)、plx4032(维罗非尼)或gsk2118436(达帕非尼)。
术语“braf抑制剂的非响应者”是指展示braf-阳性癌症的对象,其在braf抑制剂施用后显示进展,或没有或不显著的改善或癌症的治愈,或在对治疗响应一段时期后,发展出对治疗的获得性抗性。
术语“mapk/erk抑制剂”是指能够干扰mapk活性,并且特别是erk活性的分子。mapk/erk抑制剂可以是抗mapk/erk抗体,其特异性地结合mapk/erk蛋白并抑制其活性。此外,mapk/erk抑制剂可以是抑制性核酸。抑制性核酸可以是特异性结合mapk/erk蛋白并抑制其活性的适配体。其他抑制性核酸可以结合mapk/erk转录物,并抑制其翻译或使其降解。典型地,此类抑制性核酸可以是反义核酸、吗啉代寡核苷酸、抑制性rna分子,例如sirna或微小rna或核酶。此外,mapk/erk抑制剂可以是结合mapk/erk并抑制其活性的小分子。mapk/erk的此类小分子抑制剂可以通过熟知的筛选程序或分子建模方法获得,所述分子建模方法的目的是确定与mapk/erk激酶结构域的活性位点结合的化合物。一方面,本文所述的mapk/erk抑制剂是选自以下的mek抑制剂:u0126、gsk1120212(曲美替尼(trametinib))、mek162,和sch772984。更典型地,所述mapk/erk抑制剂是gsk1120212(曲美替尼(trametinib))、mek162,或sch772984。最典型地,所述mapk/erk抑制剂是erk抑制剂,并且具体为sch772984。
术语“mapk/erk抑制剂响应者”是指展示braf-阳性癌症的对象,其在mapk/erk抑制剂施用后,显示较小的进展、显著的改善或癌症的治愈。
术语“样品”是指包含癌细胞或癌细胞的蛋白和/或核酸的样品。典型地,所述癌细胞源自单细胞克隆。所述样品可源自来自肿瘤组织的生物活检材料,或体液以及得自于尸体解剖的组织。体液可以通过熟知的技术获得,并且典型地包括血液、淋巴液、肺泡、支气管或咽部灌洗液、液体或尿的样品。组织可以通过活检程序获得,其也是本领域技术人员熟知的。组织通常得自于包含肿瘤的组织并且包含癌细胞或其蛋白和/或核酸。
术语“单细胞克隆”是指亚群,并且优选在其基因组中包含braf和nras突变的癌细胞的克隆亚群。单细胞克隆可以通过本领域技术人员熟知的技术获得。此类技术通常包括从身体组织或体液分离细胞,细胞分选,并从这些单个细胞的每一个生长新的培养物。
如本文使用的术语“braf”,也称作“v-raf鼠肉瘤病毒癌基因同源物b”是指编码braf蛋白的基因。braf是raf激酶家族的成员,并且涉及影响细胞生长和分化的mapk/erk信号转导途径。braf蛋白,也被称作b-raf,在人类中是长度上由766个氨基酸组成的丝氨酸/苏氨酸激酶。其包含典型的raf激酶家族结构域:保守区1(cr1),ras-gtp-结合自调控结构域;保守区2(cr2),富丝氨酸铰链区;和保守区3(cr3),催化性蛋白激酶结构域,其使蛋白底物上的连续序列磷酸化。在其活性构象中,b-raf通过其激酶结构域的氢键合和静电相互作用形成二聚体。在本发明文中所述的braf典型地为人braf。人braf蛋白的蛋白序列已经保存在ncbi数据库中,登录号为np_004324.2,mrna/cdna序列在nm_004333.4显示(另参见seqidno:13)。小鼠braf蛋白直系同源物也是已知的,并且已保存在ncbi数据库中,登录号为np_647455.3,mrna/cdna序列在nm_139294.5显示。此术语还涵盖了上述具体braf蛋白的变体。此类变体至少具有与所述具体braf蛋白相同的基本生物和免疫学性质。具体地,如果它们可通过本说明书中所述的相同特异性测定(例如通过使用特异性识别所述braf蛋白的多克隆或单克隆抗体的elisa测定)检测,则它们共有相同的基本生物学和免疫学性质。优选的测定描述在后附的实施例中。此外,可理解根据本发明所述的变体应具有由于至少一个氨基酸取代、缺失和/或添加而不同的氨基酸序列,其中所述变体的氨基酸序列优选仍与所述具体braf蛋白的氨基酸序列具有至少50%、60%、70%、80%、85%、90%、92%、95%、97%、98%或99%的同一性。两条氨基酸序列之间的同一性程度可以通过本领域中熟知的算法测定。优选地,同一性的程度将通过在比较窗口内比较两条最优比对序列,其中比较窗口中的氨基酸序列的片段可包含与用于最佳比对的参考序列(其不包含添加或缺失)相比的添加或缺失(例如,缺口或悬端)。比较窗口优选是查询序列的完整长度,或其长度的至少50%。所述百分比的计算通过测定在两条序列中出现相同氨基酸残基的位置的数量得到匹配位置的数量,以匹配位置的数量除以比较窗口中的位置总数并将所得结果乘以100,得到序列同一性的百分比。用于比较的序列的最佳比对可以通过smith1981,add.apl.math.2:482的局部同源性算法,通过needleman1970,j.mol.biol.48:443的同源性比对算法,通过pearson1988,proc.natl.acadsci.(usa)85:2444(1988)的相似性检索法,通过这些算法的计算机化实现工具(wisconsin遗传学软件包中的gap、bestfit、blast、pasta和tfasta,geneticscomputergroup(gcg),575sciencedr.,madison,wi),或通过目视检查进行。由于已确定用于比较的两条序列,优选采用gap和bestfit测定其最佳比对,并由此测定同一性程度。优选地,使用对缺口权重为5.00,和缺口权重长度(gapweightlength)为0.30的默认值。上述变体可以是等位基因变体,或任何其他物种的特异性同源物、旁系同源物或直系同源物。此外,本文所述的变体包括特异性braf蛋白或上述类型的变体的片段,只要这些片段具有如上文所述的基本免疫性和生物性质。此类片段可以是例如braf蛋白的降解产物。进一步包括由于翻译后修饰,例如磷酸化而不同的变体。此外,上述braf蛋白可以作为单体和/或以二聚形式存在。
braf-阳性癌细胞的braf基因中的典型突变是导致braf蛋白中的一个或多个氨基酸取代的那些。一方面,所述braf蛋白中的至少一个突变是导致活化的braf蛋白的突变。另一方面,根据本发明的braf-阳性癌细胞具有突变的braf基因,其编码在对应于人braf蛋白中的氨基酸600的位置具有氨基酸取代的braf蛋白。可理解,由于在蛋白质其他位置作为诱变事件的结果而发生的氨基酸取代或添加的氨基酸,或在其他物种的旁系同源物或直系同源物中,给定氨基酸的位置可以不同。因此,如本文中所述的对应于例如人braf蛋白中位置600,即v600的位置也涵盖由于此类事件而不在600位的缬氨酸的突变,前提是所述缬氨酸的侧翼与人braf蛋白中v600的相同。加以必要的变更,这同样应用于根据本发明所述的所有其他位置编号,和对应于特异性蛋白中的特定位置的位置。在人braf基因中,氨基酸600位于外显子15中,并且由碱基对1799编码。已经在人类癌症中的所述位置确定以下氨基酸取代:缬氨酸至谷氨酸的取代(v600e)、缬氨酸至赖氨酸的取代(v600k)、缬氨酸至精氨酸的取代(v600r),或缬氨酸至天冬氨酸的取代(v600d)。因此,一方面,braf阳性细胞中的braf基因包含braf基因中的突变,其导致在对应于人braf蛋白的外显子15的氨基酸600的位置的氨基酸取代。通常,所述氨基酸取代是上述取代之一。根据本发明的braf基因可具有至少一个突变,即可具有一个或多个,例如两个,三个,四个,五个等的突变,包括上述取代之一。
如本文使用的术语“nras”也被称作“神经母细胞瘤ras病毒癌基因同源物”,如本文使用是指编码nras蛋白的基因。nras是ras激酶家族的成员,并且也涉及影响细胞生长和分化的mapk/erk信号转导途径。nras蛋白是具有固有的gtp酶活性的gtp/gdp结合蛋白。在gtp-结合阶段,其能够与raf激酶,例如braf蛋白相互作用并使其活化。在人类中,nras蛋白在长度上由189个氨基酸组成。在本发明的上下文中所述的nras典型地为人nras。人nras蛋白的蛋白序列已经保存在ncbi数据库中,登录号为np_002515.1,mrna/cdna序列在nm_002524.4显示(另参见seqidno:14)。小鼠nras蛋白直系同源物也是已知的,并且已保存在ncbi数据库中,登录号为np_035067.2,mrna/cdna序列在nm_010937.2显示。此术语还涵盖了上述特定nras蛋白的变体。此类变体至少具有与所述nras相同的基本生物和免疫学性质。具体地,如果它们可通过本说明书中所述的相同特异性测定(例如通过使用特异性识别所述nras蛋白的多克隆或单克隆抗体的elisa测定)检测,则它们共有相同的基本生物和免疫学性质。此外,可理解根据本发明所述的变体应具有由于至少一个氨基酸取代、缺失和/或添加而不同的氨基酸序列,其中所述变体的氨基酸序列优选仍与所述特定nras蛋白的氨基酸序列具有至少50%、60%、70%、80%、85%、90%、92%、95%、97%、98%或99%的同一性。两条氨基酸序列之间的同一性程度可通过本领域中熟知并且在本文他处描述的算法测定。上述变体可以是等位基因变体,或任何其他物种的特异性同源物、旁系同源物或直系同源物。此外,本文所述的变体包括特异性nras蛋白或上述类型的变体的片段,只要这些片段具有如上文所述的基本免疫性和生物性质。此类片段可以是例如nras蛋白的降解产物。进一步包括由于翻译后修饰而不同的变体。
根据本发明,所述nras基因可包含至少一个突变,即一个或多个,例如两个,三个,四个,五个等的突变。一方面,所述至少一个突变是导致nras蛋白活化的突变。另一方面,所述nras基因的突变导致在对应于人nras蛋白的外显子2的氨基酸61的位置的氨基酸取代。典型地,所述氨基酸取代是谷氨酰胺至赖氨酸的取代(q61k)、谷氨酰胺至精氨酸的取代(q61r),或谷氨酰胺至亮氨酸的取代(q61l)。在人nras基因中,氨基酸61位于外显子2中,并且由碱基对181编码。
对象的样品中至少nras基因中的至少一个突变的存在与否的测定可通过蛋白或核酸水平的各种技术进行。
在蛋白水平,突变可基于由此引出的氨基酸交换测定。为此目的,可以应用特异性检测试剂,例如特异性结合野生型(即非-突变的)或突变形式的蛋白的抗体或适配体。如果应用突变特异性检测试剂,则此类试剂的特异性结合表明存在突变,而不存在特异性结合则应表明不存在突变。
一方面,所述测定包括(i)使样品与特异性检测试剂在足以允许所述试剂与突变的nras蛋白的特异性结合的时间和条件下接触;和(ii)检测所述特异性结合的检测试剂。
如本文所述的特异性抗体优选涵盖所有类型的抗体,其优选特异性结合nras。优选地,所述抗体是单克隆抗体、多克隆抗体、单链抗体、嵌合抗体或仍能够结合nras的这些抗体的任何片段或衍生物。如本文使用的术语抗体所包括的此类片段和衍生物涵盖双特异性抗体、合成抗体、fab、f(ab)2、fv或scfv片段,或这些抗体中任一种的化学修饰的衍生物。如本发明关于抗体的上下文中使用的特异性结合是指抗体不与其他蛋白或肽交叉反应。特异性结合可以通过多种熟知的技术测试。通常,抗体或其片段可以通过使用描述于例如harlow和lane"antibodies,alaboratorymanual",cshpress,coldspringharbor,1988中的方法获得。单克隆抗体可以通过下述技术制备,其包括小鼠骨髓瘤细胞与源自免疫哺乳动物,并且优选免疫小鼠的脾细胞融合(köhler1975,nature256,495,和galfré1981,meth.enzymol.73,3)。优选地,向哺乳动物应用具有nras的突变部分的免疫原性肽。优选地,所述肽缀合至载体蛋白,例如牛血清白蛋白、甲状腺球蛋白和钥孔血蓝蛋白(klh)。取决于宿主的物种,可使用各种佐剂以提高免疫反应。优选地,此类佐剂涵盖弗氏佐剂、矿物凝胶,例如氢氧化铝和表面活性物质,例如溶血卵磷脂、普朗尼克多元醇(pluronicpolyols)、聚阴离子、肽、油乳液、、匙孔血蓝蛋白和二硝基苯酚。随后,可以使用熟知的杂交瘤技术,人b细胞杂交瘤技术和ebv杂交瘤技术制备特异性结合b-型丛蛋白(b-typeplexin)的胞外结构域的单克隆抗体。
优选地,如本文使用的特异性适配体是结合特异性靶分子的寡核酸或肽分子(ellington1990,nature346(6287):818–22).bock1992,nature355(6360):564–6)。通过重复的选择轮次或所谓通过指数富集的配体的系统演变(selex技术),对寡核酸适配体进行工程化。肽适配体经设计用于干扰细胞内的蛋白质相互作用。它们通常包含在两个末端都连接至蛋白质骨架的可变肽环。这种双结构约束将肽适配体的结合亲和力提高至纳摩尔范围。优选地,所述可变肽环的长度是由10至20个氨基酸构成,并且所述骨架可以是具有改善的溶解性和强度(compacity)性质的任何蛋白,例如硫氧还蛋白-a。肽适配体选择可以使用不同的系统,包括例如酵母双杂交系统(参见例如hoppe-seyler2000,.jmolmed.78(8):426–30)进行。
特异性抗体或适配体可以连接至可检测的标记。适合的可检测标记包括金颗粒、乳胶珠、二氢吖啶酯、鲁米诺、钌、酶活性标记、放射性标记,磁性标记(“例如磁珠”,包括顺磁性和超顺磁性标记),和荧光标记。酶活性标记包括例如辣根过氧化物酶、碱性磷酸酶、β-半乳糖苷酶、荧光素酶及其衍生物。用于检测的适合底物包括二氨基-联苯胺(dab)、3,3'-5,5'-四甲基联苯胺、nbt-bcip(4-硝基蓝四唑氯化物和5-溴-4-氯-3-吲哚基-磷酸盐,其可作为现成储备溶液得自rochediagnostics),cdp-star™(amershambiosciences),ecf™(amershambiosciences)。适合的酶-底物组合可导致有色的反应产物,荧光或化学发光,其可根据本领域中已知的方法测量(例如,使用光敏胶片或适合的摄像系统)。典型的荧光标记包括荧光蛋白(例如gfp及其衍生物bfp、rfp等),肽标签,例如his-标签、flag-标签、myc-标签等,cy3、cy5、德州红(texasred)、荧光素,和alexa染料(例如alexa568)。进一步的荧光标记可得自于例如molecularprobes(oregon)。还预期到量子点作为荧光标记的用途。典型的放射性标记包括35s、125i、32p、33p等。
上述标记的存在与否可通过本领域中熟知的方法和设备测试,包括生物传感器、与免疫测定偶联的光学设备,分析设备,例如质谱仪、nmr-分析仪,或层析设备。进一步地,方法包括基于elisa(酶联免疫吸附测定)的方法,全自动或机器人的免疫测定,例如可在elecsystm分析仪上获得;cba,其为酶促钴结合测定,可例如在roche-hitachitm分析仪上获得;以及乳胶凝集测定,例如可在roche-hitachitm分析仪上获得。根据本发明适合的测量方法还包括沉淀法,特别是免疫沉淀;电化学发光、ria(放射免疫测定)、夹心酶免疫测试、电化学发光夹心免疫测定(eclia)、解离增强镧系荧光免疫测定(delfia)、闪烁迫近测定(spa)、比浊法,浊度测定、乳胶增强比浊法或浊度测定,或固相免疫测试。本领域中已知的其他方法,例如凝胶电泳、2d凝胶电泳、sds聚丙烯酰胺凝胶电泳(sds-page)和western印迹,可以单独使用或与如上文所述的标记或其他检测方法组合使用。
另一方面,可以直接检测突变的nras蛋白。为此目的,可以通过质谱或基于nmr的技术测量物理或化学性质中的差异。可选地,可以测量生物活性中的差异,例如在无细胞或基于细胞的测试系统(活性测试)中提高的生物活性。
在核酸水平,可以通过测定基因或其编码蛋白的转录物的核酸序列,来测量突变。为此目的,可以应用特异性结合野生型(即非突变)或突变形式的基因或其转录物的核酸或寡核苷酸。如果应用突变特异性核酸或寡核苷酸,则此类试剂与基因或其转录物或其扩增子的特异性结合表明存在突变,而缺少特异性结合则应表明不存在突变。
一方面,所述测定包括(i)使样品与特异性核酸或寡核苷酸在足以允许所述试剂与突变的nras基因或其转录物的特异性结合的时间和条件下接触;和(ii)检测所述特异性结合的核酸或寡核苷酸。通常,根据本发明的这个方面应用杂交技术。所述杂交技术包括southern印迹杂交或northern印迹杂交。
另一方面,所述测定包括(i)使样品与特异性引物寡核苷酸在足以允许特异性扩增所述突变nras基因的一部分的时间和条件下接触,其中所述特异性引物寡核苷酸仅允许所述突变nras基因的扩增;和(ii)检测所述扩增产物。在这个方面,扩增产物的存在表明存在突变的nras基因,而缺少扩增产物则表明不存在突变的nras基因。通常,根据本发明的这个方面应用基于pcr的技术。所述基于pcr的技术包括pcr、rt-pcr、巢式pcr、qpcr、荧光定量pcr(lightcyclepcr)、实时pcr、irt-pcr、触底-pcr(touchdown-pcr)、多重pcr、数字pcr等。
在进一步方面,所述测定包括对突变的nras或其转录物,特别是突变的一个或多个碱基对进行测序。通常,可应用根据sanger或maxam-gilbert的常规测序。可选地,可应用先进测序技术例如鸟枪测序、桥式pcr、大规模平行特征测序(mpss)、polony测序、454焦磷酸测序、illumina(solexa)测序、solid测序,iontorrent半导体测序、dna纳米球测序、heliscope单分子测序、单分子实时(smrt)测序、纳米孔dna测序、隧道电流dna测序、杂交测序、质谱测序、微流sanger测序,基于显微镜的技术和rnap测序。
更典型地,通过基于杂交的技术测定nras核酸的催化亚基的外显子2中的所述至少一个突变的存在,并且具体地通过:
a)使来自所述对象的样品中的核酸接触一种或多种选自以下的基因座特异性寡核苷酸:ggtgaaacctgtttgttggacat(seqidno:7);tgtattggtctctcatggcactgt(seqidno:8);gataggcagaaatgggcttga(seqidno:9);和atcatcctttcagagaaaataatgc(seqidno:10);
b)将所述样品在允许所述寡核苷酸与其在nras核酸中的靶序列的特异性杂交的条件下温育;
c)检测所述杂交;和
d)基于步骤c)中检测的所述杂交测定所述至少一个突变。
进行接触,从而使得所述一个或多个基因座特异性寡核苷酸可以在物理上接近待检测的核酸,即编码具有所述至少一个突变的nras蛋白的核酸(nras核酸)。
本领域技术人员能够测定在存在突变的情况下,仅允许所述一个或多个基因座特异性寡核苷酸与nras核酸中的nras靶序列的杂交的特异性杂交条件,而无需再费周折。所述条件可取决于所应用的一个或多个基因座特异性寡核苷酸而改变。具体设想的条件是下文所附实施例中所述的那些。
特异性杂交的检测可以通过任何技术进行,所述技术允许检测所述基因座特异性寡核苷酸和所述靶核酸的核酸杂交体。通常,所述基因座特异性寡核苷酸可以偶联至可检测的标记。在杂交技术的背景下,用于核酸的适合的可检测标记是本领域中熟知的,并且涵盖例如放射性标记、荧光标记、显色标记、染料酶标记、可通过抗体或适配体检测的标记等。具体设想的标记是下文所附实施例中所述的那些。
所述至少一个突变的测定通过检测特异性杂交来进行。关于寡核苷酸的基因座特异性的信息进一步表明通过杂交检测的突变的类型,即由于所述寡核苷酸已设计用于与包含例如特定突变的特定靶序列杂交,所以检测的杂交也表明所述特定突变在靶核酸中的存在。
典型地,步骤b)进一步包括生成包含nras核酸内的靶序列的扩增产物的步骤,其通过使用以下寡核苷酸引物中的一个或两个扩增样品中的nras核酸:具有seqidno:11的正向寡核苷酸引物和具有seqidno:12的反向寡核苷酸引物。
可以通过具体如本文他处所述的pcr进行扩增,即所述正向和反向引物允许退火至靶序列,从而可以发生dna合成。随后,分离新合成的dna链,并再次开启循环。典型地,所述扩增pcr进行15-45个循环,更典型为16-40个循环,并且甚至更典型地进行16-30个循环。适合的pcr条件取决于所应用的正向和反向引物,并且可以通过本领域技术人员确定,而无需再费周章。根据本发明设想的具体pcr条件是后附实施例中所述的那些。
为了进一步增强通过本发明方法进行的评估,还设想nras之外的其他癌症生物标记物。一方面,所述方法进一步涵盖测定braf基因中至少一个突变的存在与否,由此所述至少一个突变的存在进一步将所述对象确定为braf抑制剂非响应者和mapk/erk抑制剂响应者。待测定的所述至少一个braf突变通常是上文所述的braf氨基酸取代之一。也可以与本文他处所述的至少一个nras突变的测定类似的方式,在蛋白或核酸水平测定所述braf突变。
更典型地,通过基于杂交的技术测定braf核酸的催化亚基的外显子15中的所述至少一个突变的存在,并且具体地通过:
a)使来自所述对象的样品中的核酸接触一种或多种选自以下的基因座特异性寡核苷酸:ctaagaggaaagatgaagtactatg(seqidno:1);ctagtaactcagcagcatctcag(seqidno:2);ctactgttttcctttacttactacacctcaga(seqidno:3);和atccagacaactgttcaaactgat(seqidno:4);
b)在允许所述寡核苷酸与其在braf核酸中的靶序列的特异性杂交的条件下温育所述样品;
c)检测所述杂交;和
d)基于步骤c)中检测的所述杂交测定所述至少一个突变。
进行接触,从而使得所述一个或多个基因座特异性寡核苷酸可以在物理上接近待检测的核酸,即编码具有所述至少一个突变的braf蛋白的核酸(braf核酸)。
本领域技术人员能够测定在存在突变的情况下,仅允许所述一个或多个基因座特异性寡核苷酸与braf核酸中的braf靶序列的杂交的特异性杂交条件,而无需再费周折。所述条件可取决于所应用的一个或多个基因座特异性寡核苷酸而改变。具体设想的条件是下文所附实施例中所述的那些。
特异性杂交的检测可以通过任何技术进行,所述技术允许检测所述基因座特异性寡核苷酸和所述靶核酸的核酸杂交体。通常,所述基因座特异性寡核苷酸可以偶联至可检测的标记。具体设想的标记是下文所附实施例中所述的那些。
所述至少一个突变的测定通过检测特异性杂交来进行。关于寡核苷酸的基因座特异性的信息进一步表明通过杂交检测的突变的类型,即由于所述寡核苷酸已设计用于与包含例如特定突变的特定靶序列杂交,所以检测的杂交也表明所述特定突变在靶核酸中的存在。
典型地,步骤b)进一步包括生成包含braf核酸内的靶序列的扩增产物的步骤,其通过使用以下寡核苷酸引物中的一个或两个扩增样品中的nras核酸:具有seqidno:5的正向寡核苷酸引物和具有seqidno:6的反向寡核苷酸引物。
所述扩增可以通过pcr进行,如本文他处所述。根据本发明设想的具体pcr条件是后附实施例中所述的那些。
如果已经如上文所述确定nras基因中的所述至少一个突变,则将对象确定为braf抑制剂非响应者和mapk/erk抑制剂响应者。通常,所述确定将导致向所述对象推荐待应用的治疗措施。如本文他处所讨论的,发现根据本发明具有在nras蛋白中的至少一个突变的对象将会是braf抑制剂非响应者,但同时将响应mapk/erk抑制剂。因此,根据本发明设想基于适当的确定,可给予这种对象适当疗法的推荐。因此,一方面,本发明的方法进一步包括如果对象被确定为braf抑制剂非响应者和mapk/erk抑制剂响应者,则向所述对象推荐mapk/erk抑制剂的施用,特别是如本文所述的mapk/erk抑制剂。另一方面,所述方法可进一步包括向所述对象施用所述mapk/erk抑制剂,特别是如本文所述的mapk/erk抑制剂,并且,在另一方面,调整braf抑制剂施用的剂量或停止braf抑制剂的施用,特别是本文所述的braf抑制剂。
为了更好地表征在不同治疗方式下患者间异质性的进化,在本发明的基本研究中,对来自分别接受不同疗法但在治疗下快速进展的3名iv期黑素瘤患者的多个样品进行外显子组(exome)测序。所使用的来自不同阶段(取决于可得性)剩余活检材料包括血液、发育不良痣、原发肿瘤,治疗之前的转移瘤和死亡后尸检期间获得的转移瘤。为了更好地表征肿瘤内异质性,在可能的情况下对相同原发肿瘤的多个组织学上不同的区域进行测序,并从用于靶向再测序的早期传代培养物制备单细胞克隆。特异性靶向逐渐提高的途径抑制剂供应线与强力的下一代测序的应用的组合有利地允许改进的表征和治疗方案,其经调整针对与转移性黑素瘤进展最相关的关键驱动途径2,22,23。
特别地,为了更好地表征癌症患者个体如何响应标准治疗,确定了具有相似治疗时间进程,但不同的致癌突变和治疗方案的三名患者。首位患者具有brafv600e突变,并且具有对靶向性braf抑制剂治疗的初始响应。患者2对braf和nras均为纯合野生型,并且接受帕唑帕尼,其为多受体酪氨酸激酶抑制剂。最后,患者3具有nrasq61r突变,并施用了mek抑制剂。从由多个活检获得的ffpe材料钻孔产生完整外显子组测序数据,并参考从各个患者血液分离的种系dna。与研究更大患者组群,但来自每位患者的样品较少的早期研究相比,这种方法提供了患者间基因组异质性的更全面的见解。
通过分析存在于患者肿瘤中的高质量单核苷酸变异(snv),可显示各患者的原发肿瘤包含与其所有转移瘤相比最大的遗传多样性。这与以下预期相符,即癌症起源位点会比后代包含更多的遗传变异,所述后代在后来出现并且推测具有较少的用于获得从头突变的时间。有趣的是,来自患者1的两个发育异常的痣都具有比从所述单位患者测序的任何肿瘤样品更低的蛋白质编码突变负担。尽管这一点的原因尚不清楚,所述痣的遗传多样性降低可能是基因组不稳定性较低的结果,或者可能是累积突变的时间段较短,以及其他可能的原因。
进一步使用这些数据的完整外显子组系统发生分析推断各患者内肿瘤之间的进化关系,并测定每种治疗方案如何影响遗传异质性的进化。不同于显示在靶向治疗后克隆的分支进化的此前研究,可见在braf和mek抑制剂二者治疗后,转移瘤的强烈的良好支持的单系演化出现并复发。与之相比,接受多激酶抑制剂(即帕唑帕尼)的患者2不具有晚期肿瘤转移的单系布局,表明晚期转移瘤之间的遗传漂变。
有趣的是,尽管接受braf抑制剂的患者中有晚期转移瘤的单系分离,但在所有测序的活检样品之间没有共有的已知抗性机制。实际上,通过sanger测序和数字pcr都确定活化突变nrasq61k存在于患者1的单个转移瘤中,但不存在于来自该患者的所有其他抗性肿瘤样品中。这与此前公布的显示患者个体内的抗性机制的异质性的数据相吻合11,并且使得编目病因和治疗已发展出治疗抗性患者的工作均更为艰巨。因此,尽管我们观察到治疗后亚克隆的单系选择,但不同的转移瘤可能包含不同的抗性机制。
通过分离源自这种抗性肿瘤的26个单细胞克隆的群落并测序,可首次显示在单个肿瘤细胞中存在两种活化mapk突变。在正常培养条件下生长的这些双突变细胞对于已用于治疗患者的braf抑制剂具有抗性,但对两种其他braf抑制剂仅具有部分抗性。尽管这些细胞保持对braf抑制的抗性,但在lgx818和plx4032存在下仍可观察到perk水平降低。重要的是,双突变细胞仍对组合的mek和braf抑制,以及单剂mek和erk抑制敏感。这种观察结果表明尽管存在赋予抗性的突变,但使用其他mapk-途径抑制剂,并且特别是mapk/erk抑制剂的同时治疗或二线治疗,仍可有效地控制进展。
然而,由于双突变基因型仅存在于患者1的其他5个转移瘤中的晚期转移瘤#6,并且赋予其他肿瘤治疗抗性的基本机制尚不清楚,这些二线或组合治疗在控制总体肿瘤负担中的功效尚存有疑问。如果患者1中其他肿瘤活化了不同的途径,例如pi3k,pten和akt,从而导致其对mapk抑制不敏感,尤其将是如此。通过数字pcr,证实甚至在单个抗性肿瘤中,双突变细胞的频率也是可变的,表明这些细胞也可以旁分泌的方式促成抗性,或者可具有抗性机制中的肿瘤内异质性。
在本发明的基本研究中展示的接受靶向抑制的患者中的癌细胞的单系进化,表明对可更好地在该治疗环境下存活的异质亚克隆的选择。然而,这些肿瘤之间明显缺少共同的抗性机制,表明随后出现的抗性可通过我们的方案无法确定的共有遗传机制发生,通过非遗传途径发生,或在每个转移瘤个体中不同的方式发生。所有这些可能性都提出了严重的治疗挑战。但是,双突变黑素瘤细胞对mapk-抑制的剩余敏感性表明如果考虑存在于转移性黑素瘤患者中的空间和时间遗传异质性,在精密医药的背景下,使用mapk-途径抑制剂的组合和二线疗法代替或附加例如braf抑制剂仍可以是有效的。
由于本发明,现在可以表征癌症并且特别是具有braf-阳性癌细胞的癌症对braf抑制剂的抗性,并为那些抗性患者选择更有效的疗法。此外,本发明还提供了基于mek/erk抑制剂在患者中的使用的更有效的疗法,所述患者患有显示对braf抑制剂抗性的braf-阳性癌症。大体上,本发明的基本研究还提供了用于诊断或评估癌症,特别是关于携带至少一个nras和至少一个braf突变的双突变癌细胞的诊断方法。
加以必要的变更,上文对术语的定义解释应用于以下实施方式。
以下描述了本发明的典型实施方式:
在本发明方法的一个实施方式中,所述方法进一步包括测定braf基因中至少一个突变的存在与否,由此所述至少一个突变的存在进一步将所述对象确定为braf抑制剂非响应者和mapk/erk抑制剂响应者。
在本发明方法的另一实施方式中,所述braf-阳性癌症是黑素瘤癌。
在本发明方法的进一步实施方式中,所述braf-阳性癌症由源自单细胞克隆的细胞群组成。
在本发明方法的又一实施方式中,所述细胞群的细胞在其基因组中包含在braf基因中的至少一个突变和在nras基因中的至少一个突变。
在本发明方法的又一实施方式中,所述braf抑制剂是braf活性的小分子抑制剂。典型地,所述braf活性的小分子抑制剂是lgx818、plx4032和/或gsk2118436。
在本发明方法的又一实施方式中,所述mapk/erk抑制剂是mek或erk活性的小分子抑制剂。典型地,所述mek活性的抑制剂是gsk1120212或mek162,并且所述erk活性的抑制剂是sch772984。
在本发明方法的又一实施方式中,所述nras基因的突变导致在对应于人nras蛋白的外显子2的氨基酸61的位置的氨基酸取代。典型地,所述氨基酸取代是谷氨酰胺至赖氨酸的取代(q61k)、谷氨酰胺至精氨酸的取代(q61r),或谷氨酰胺至亮氨酸的取代(q61l)。
在本发明方法的进一步实施方式中,所述braf基因的突变导致在对应于人braf蛋白的外显子15的氨基酸600的位置的氨基酸取代。典型地,所述氨基酸取代是缬氨酸至谷氨酸的取代(v600e)、缬氨酸至赖氨酸的取代(v600k)、缬氨酸至精氨酸的取代(v600r),或缬氨酸至天冬氨酸的取代(v600d)。
在本发明方法的又一实施方式中,所述样品包含braf-阳性癌细胞。
在本发明方法的进一步实施方式中,所述样品选自:组织切除样品、组织活检样品、原发肿瘤样品、转移性损伤的样品或包含循环肿瘤细胞的样品(包括血液)。
在本发明方法的实施方式中,通过以下测定nras核酸的催化亚基的外显子2中的所述至少一个突变的存在:
a)使来自所述对象的样品中的核酸接触一种或多种选自以下的基因座特异性寡核苷酸:ggtgaaacctgtttgttggacat(seqidno:7);tgtattggtctctcatggcactgt(seqidno:8);gataggcagaaatgggcttga(seqidno:9);和atcatcctttcagagaaaataatgc(seqidno:10);
b)在允许所述寡核苷酸与其在nras核酸中的靶序列的特异性杂交的条件下温育所述样品;
c)检测所述杂交;和
d)基于步骤c)中检测的所述杂交测定所述至少一个突变。
典型地,步骤b)进一步包括生成包含nras核酸内的靶序列的扩增产物的步骤,其通过使用以下寡核苷酸引物中的一个或两个扩增样品中的nras核酸:具有seqidno:11的正向寡核苷酸引物和具有seqidno:12的反向寡核苷酸引物。
在本发明方法的实施方式中,通过以下测定braf核酸的催化亚基的外显子15中的所述至少一个突变的存在:
a)使来自所述对象的样品中的核酸接触一种或多种选自以下的基因座特异性寡核苷酸:ctaagaggaaagatgaagtactatg(seqidno:1);ctagtaactcagcagcatctcag(seqidno:2);ctactgttttcctttacttactacacctcaga(seqidno:3);和atccagacaactgttcaaactgat(seqidno:4);
b)在允许所述寡核苷酸与其在braf核酸中的靶序列的特异性杂交的条件下温育所述样品;
c)检测所述杂交;和
d)基于步骤c)中检测的所述杂交测定所述至少一个突变。
典型地,步骤b)进一步包括生成包含braf核酸内的靶序列的扩增产物的步骤,其通过使用以下寡核苷酸引物中的一个或两个扩增样品中的nras核酸:具有seqidno:5的正向寡核苷酸引物和具有seqidno:6的反向寡核苷酸引物。
在本发明方法的又一实施方式中,所述方法进一步包括如果所述对象已经被确定为braf抑制剂非响应者和mapk/erk抑制剂响应者,则向所述对象推荐mapk/erk抑制剂药物的施用。
本发明还涉及mapk/erk抑制剂,其用于治疗患有braf-阳性癌症的对象,由此已发现所述癌症:(i)至少具有在nras基因中的至少一个突变,或(ii)至少具有在nras基因中的至少一个突变和在braf基因中的至少一个突变。此外,根据本发明预期了mapk/erk抑制剂在用于治疗braf-阳性癌症患者的药物的制备中的用途,由此已发现所述癌症:(i)至少具有在nras基因中的至少一个突变,或(ii)至少具有在nras基因中的至少一个突变和在braf基因中的至少一个突变。
因此,所述mapk/erk抑制剂将作为药物用于治疗,并因此可如此配制。一方面,如本文使用的术语“药物”是指包含上文所述药物活性化合物的抑制剂的药物组合物,其中所述药物组合物可以治疗有效的剂量用于人或非人的本文指定的疾病的治疗。通常,所述抑制剂可以液体或冻干形式存在。一方面,所述药物用于局部或系统施用。常规地,药物将在肌肉内或皮下施用。然而,取决于化合物的性质和作用模式,所述药物也可通过其他途径施用。所述抑制剂应为组合物的活性成分,并且通常以常规剂型施用,所述剂型通过根据常规方法将药物与标准药物载体组合而制备。这些方法可涉及根据需要将所述成分混合、粒化和压缩,或溶解成期望的制剂。可理解,药学可接受载体或稀释剂的形式和特征受控于将预期组合的活性成分的量、施用途径和其他熟知变量。载体必须在与制剂的其他成分相容以及不对其受体有害方面是可接受的。所采用的药物载体可包括固体、凝胶或液体。固体载体的实例为乳糖、石膏粉、蔗糖、滑石粉、明胶、琼脂、果胶、阿拉伯胶、硬脂酸镁、硬脂酸等。液体载体的实例为磷酸缓冲盐水溶液、糖浆、油、水、乳剂、各种类型的润湿剂等。类似地,所述载体或稀释剂可包括本领域熟知的时间延迟材料,例如单独或与蜡一起的单硬脂酸甘油酯或二硬脂酸甘油酯。所述适合的载体包括上文所述的那些和本领域熟知的其他载体,参见例如remington'spharmaceuticalsciences,mackpublishingcompany,easton,pennsylvania。对稀释剂进行选择以免影响组合的生物活性。此类稀释剂的实例为蒸馏水、生理盐水、林格氏溶液、葡萄糖溶液和汉克氏溶液。此外,所述药物组合物或制剂也可包括其他载体、佐剂,或非毒性非治疗性非免疫原性稳定剂等。治疗上有效的剂量是指根据本发明将用于药物中的化合物的量,其防止、改善或治疗伴随本说明书中所述疾病的症状。化合物的治疗功效和毒性可以通过标准药物程序在细胞培养物或实验动物中测定,例如ed50(在50%的群体中治疗有效的剂量)和ld50(对50%的群体致死的剂量)。治疗和毒性作用之间的剂量比例是治疗指数,并且可将其表示为ld50/ed50比例。剂量方案将由主治医师和临床因素决定。如医药领域中熟知的,对任一患者的剂量取决于很多因素,包括患者的体型、体表面积、年龄、待施用的具体化合物、性别、施用的时间和途径,一般健康状况,以及同时施用的其他药物。可通过周期性评估监测进展。本文所指的药物施用至少一次,以治疗或改善或预防本说明书中所述的疾病或病症。然而,所述药物可以施用多于一次。具体药物按照制药领域中熟知的方式制备,并且包含上文所述的至少一种活性化合物,其与药学上可接受的载体或稀释剂混合或结合。为了制备这些具体药物组合物,通常将所述一种或多种活性化合物与载体或稀释剂混合。所得制剂要适应施用模式。剂量推荐应标明在处方或使用说明中,以预见取决于所考虑的受体的剂量调整。在另一方面,本发明的根据本发明的药物包含在其配制期间添加到所述药物的mapk/erk抑制剂之外的药物。此类药物的细节可见于本文他处。最后,可理解药物的配制在gmp标准化条件等下进行,以确保药物的质量、药物安全和有效性。
根据上文,所述mapk/erk抑制剂也可用于治疗患有braf-阳性癌症的对象的braf-阳性癌症的方法,所述方法包括向所述对象施用治疗有效量的mapk/erk抑制剂。
本发明还涉及在疑似患有癌症的对象的样品中诊断癌症的方法,其包括:
a)产生包含braf核酸和nras核酸内的靶序列的一种或多种扩增产物,其中使用以下引物寡核苷酸中的两条:ctaagaggaaagatgaagtactatg(seqidno:1);ctagtaactcagcagcatctcag(seqidno:2);ctactgttttcctttacttactacacctcaga(seqidno:3);和/或atccagacaactgttcaaactgat(seqidno:4),和以下引物寡核苷酸中的两条扩增样品中的核酸:ggtgaaacctgtttgttggacat(seqidno:7);tgtattggtctctcatggcactgt(seqidno:8);gataggcagaaatgggcttga(seqidno:9);和/或atcatcctttcagagaaaataatgc(seqidno:10);
b)使所述核酸样品接触以下突变特异性braf寡核苷酸中的一条或多条:ctaagaggaaagatgaagtactatg(seqidno:1);ctagtaactcagcagcatctcag(seqidno:2);ctactgttttcctttacttactacacctcaga(seqidno:3);和/或atccagacaactgttcaaactgat(seqidno:4);
以及以下位置特异性nras寡核苷酸中的一条或多条:ggtgaaacctgtttgttggacat(seqidno:7);tgtattggtctctcatggcactgt(seqidno:8);gataggcagaaatgggcttga(seqidno:9);和/或atcatcctttcagagaaaataatgc(seqidno:10);
c)在允许所述寡核苷酸与其各自在braf核酸和nras核酸内的靶序列的特异性杂交的条件下温育所述样品;
d)检测所述杂交,从而诊断癌症。
如本文使用的术语“诊断”是指评估本文所述的对象患有癌症(即归入癌症患者组),还是没患癌症(即,排除)。如本领域中那些技术人员所理解的,此类评估通常并非意在对于100%的待诊断对象都是正确的。然而,此术语需要是否存在癌症的评估对统计学上显著部分的对象(例如,群组研究中的群组)是准确的。可如本文他处所述确定某部分是否是统计学上显著的。
如本文使用的术语“癌症”是指由异常细胞生长和侵袭表征的所有恶性肿瘤。具体地,本文所指的癌症是如本文他处所述的braf-阳性癌症。
如本文所述的短语“产生一种或多种扩增产物”可以通过任何基于引物的核酸扩增技术实现。一方面,通过本文他处或在后附实施例中详述的基于pcr的技术实现所述产生。
在上述方法的实施方式中,所述癌症源自单细胞克隆。
本发明还涵盖用于在对象的样品中诊断癌症,通常是源自单细胞克隆的癌症的试剂盒,其包含以下寡核苷酸:ctaagaggaaagatgaagtactatg(seqidno:1);ctagtaactcagcagcatctcag(seqidno:2);ctactgttttcctttacttactacacctcaga(seqidno:3);atccagacaactgttcaaactgat(seqidno:4);ggtgaaacctgtttgttggacat(seqidno:7);tgtattggtctctcatggcactgt(seqidno:8);gataggcagaaatgggcttga(seqidno:9);和atcatcctttcagagaaaataatgc(seqidno:10)。
如本文使用的术语“试剂盒”是指上述成分的集合,典型地,其分离地或在单个容器内提供。所述容器还包含用于实施本发明方法的说明书。这些说明书可以是手册的形式,或者通过计算机程序代码提供,所述计算机程序代码能够进行本发明方法所述的确定,并且在计算机或数据处理设备上执行时建立诊断。所述计算机程序代码可提供在数据存储介质或设备上,例如光存储介质(例如压缩盘),或直接提供在计算机或数据处理设备上。进一步地,所述试剂盒可包含阳性和阴性对照靶核酸。一方面,所述试剂盒还可包含实施本发明方法所需的其他成分,例如检测试剂,例如抗体,缓冲剂,检测所需的其他试剂,例如缀合物和/或底物等。
本发明进一步涵盖用于在疑似患有癌症的对象的样品中诊断癌症,通常为源自单细胞克隆的癌症,和/或用于确定患有braf-阳性癌症的对象是否为braf抑制剂非响应者和/或为mapk/erk的抑制剂响应者的设备,其包含:
(i)分析单元,其包含以下突变特异性braf寡核苷酸的一种或多种:ctaagaggaaagatgaagtactatg(seqidno:1);ctagtaactcagcagcatctcag(seqidno:2);ctactgttttcctttacttactacacctcaga(seqidno:3);和/或atccagacaactgttcaaactgat(seqidno:4),以及以下位置特异性nras寡核苷酸的一种或多种:ggtgaaacctgtttgttggacat(seqidno:7);tgtattggtctctcatggcactgt(seqidno:8);gataggcagaaatgggcttga(seqidno:9);和/或atcatcctttcagagaaaataatgc(seqidno:10);和
(ii)检测器,其能够检测braf和nras核酸与所述寡核苷酸的特异性杂交。
如本文使用的术语“设备”是指包含彼此可操作连接的上述成分,以允许根据本发明方法的诊断或确定的系统。一方面,所述分析单元包含在固体支持物上的固定形式的所述寡核苷酸,其将与包含待测定的靶核酸的样品接触。所述分析单元可进一步包含或可操作地连接至包含用于实施所述杂交反应的清洗或杂交溶液的容器。
所述检测器适合用于检测所述寡核苷酸和所述靶核酸的特异性杂交。取决于用于所述寡核苷酸的标记,可使用不同的检测器,例如光学检测器可在荧光标记或染料的情况下应用。
所述设备可进一步包括用于数据计算的计算设备。例如,计算设备可以是通用计算机或便携式计算设备。还应理解,可将多个计算设备一起使用,例如经网络或传输数据的其他方法,用于实施本文公开方法的一个或多个步骤。示例性的计算设备包括台式计算机、膝上型计算机、个人数据助理和智能电话、蜂窝设备、平板计算机、服务器等。通常,计算设备包含能够执行多个指令(例如软件的程序)的处理器。计算设备可访问内存。内存是计算机可读介质,并且可包含单个存储设备或多个存储设备,或与计算设备一起位于本地或可例如跨网络访问计算设备。计算机可读介质可以是任何可得的介质,其能够通过计算设备访问,并包括易失性和非易失性介质。进一步地,计算机可读介质可以是可移动和不可移动介质中的一者或二者。例如,计算机可读介质可包含计算机存储介质,但不限于此。示例性的计算机存储介质包括但不限于ram、rom、eeprom、闪存或任何其他内存技术,cd-rom、数字通用盘(dvd)或其他光盘存储器、磁带盒、磁带、磁盘存储器或其它磁存储设备,或任何其他介质,其能够用于存储能够通过所述计算设备访问并通过所述计算设备的处理器执行的多个指令。
所述计算设备也可访问输出设备。示例性的输出设备包括例如传真机、显示器、打印机和文件。根据本公开的一些实施方式,计算设备可执行本文公开的方法的一个或多个步骤,并且此后通过输出设备提供涉及本发明结果的输出。
本发明设想了评估患者针对典型地来自单细胞克隆的癌症的靶向治疗的响应性的方法,其包括:
a)提供来自所述患者的样品,
b)测试所述样品中braf和nras基因中的突变的存在,其中通过选自:选择性扩增、探针杂交或核酸测序的方法之一进行所述测试;
c)如果检测到braf和nras基因中的突变,检测对mapk/erk抑制剂的响应性以及对braf抑制剂的非响应性。
在上述方法的一方面,通过如本文他处详述的选择性扩增、探针杂交或核酸测序测定braf和nras基因中的突变的存在。具体地,可使用本文他处指定的基因座特异性或突变特异性寡核苷酸或引物寡核苷酸。
此外,本发明涉及评估患者中典型地源自单细胞克隆的癌症的方法,其包括:
a)提供或获得来自所述患者的包含核酸的样品,
b)使所述样品中的核酸接触对braf和nras基因中的突变特异性的核酸探针,
c)如果检测到braf和nras基因中的突变,将所述癌症评估为响应mapk/erk抑制剂且不响应braf抑制剂。
又进一步地,本发明涉及评估患者中典型地源自单细胞克隆的癌症的方法,其包括:
a)提供或获得来自所述患者的包含核酸的样品,
b)使所述样品中的核酸接触对braf和nras基因中的突变特异性的核酸探针,
c)如果检测到braf和nras基因中的突变,报告所述癌症为响应mapk/erk抑制剂且不响应braf抑制剂。
在上述方法的一个方面,对braf和nras基因中的突变特异性的核酸探针是本文他处指定的基因座特异性或突变特异性寡核苷酸或引物寡核苷酸。
本说明书全文引用的所有文献就其具体提及的公开内容及其全文通过引用并入本文。
附图
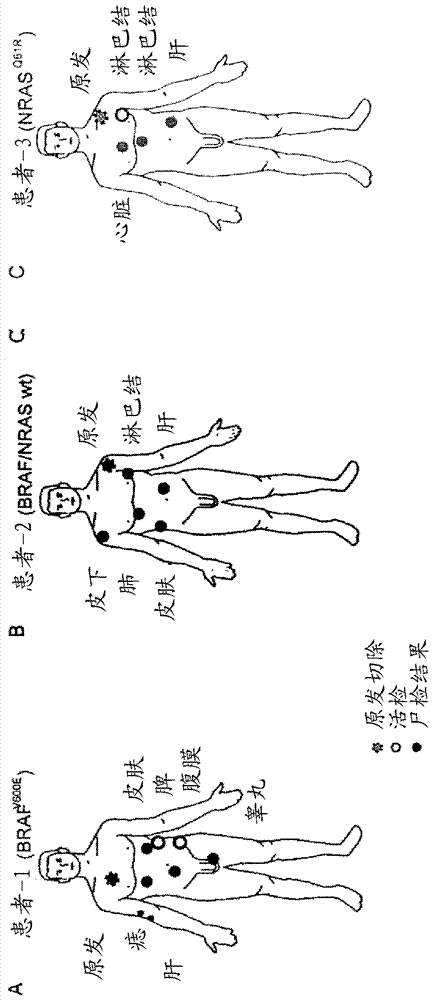
图1:患者组群和拷贝数变异。(a)来自患者1的样品包括原发肿瘤,两个发育异常的痣,肿瘤复发后的两个早期转移瘤和4个晚期转移瘤。(b)患者1具有brafv600e突变的黑素瘤,并且先后接受ifna治疗和特异性braf抑制剂治疗,他对此做出响应,但随后变为抗性的。(c)患者2被诊断为患有对braf和nras均为野生型的黑素瘤。对原发肿瘤钻孔取样并测序三次。对另外5个晚期转移瘤进行测序。(d)患者接受了多受体酪氨酸激酶抑制剂(帕唑帕尼),他对此做出响应但随后变为抗性的。(e)患者3具有nrasq61r突变。对原发肿瘤钻孔取样两次,并从一个早期和三个晚期转移瘤获取活检样品。(f)患者接受了mek抑制剂gsk1120212,他对此做出响应但随后在短期抗ctla4治疗后变为抗性的。(g)使用circos绘制拷贝数变化(cnv)。每个环显示通过一个活检样品的钻孔检测的cnv,从两个最外圈中的两个痣开始,随后为原发肿瘤,两个早期转移瘤,并且随后为晚期转移瘤1至4。(h)展示了患者2的cnv,从外向内依次为:原发肿瘤样品1至3和晚期转移瘤1至5。(i)显示了患者3的cnv,从外向内依次为:原发肿瘤样品1和2,一个早期转移瘤和晚期转移瘤1至3。放大区域显示染色体9中常丢失的区域,其编码肿瘤抑制因子cdkn2a。(k)患者1的染色体22中的拷贝数变化显示高度的异质性。原发肿瘤具有22p区域中的增加,以及在22p和22q的大区域中的丢失。所述增加(而不是丢失)可见于早期转移瘤1,但在其他转移瘤中则没有发现。所述丢失(而不是增加)可见于早期转移瘤2和晚期转移瘤1,但在其他转移瘤中则没有发现。
图2:患者活检样品的全外显子组系统发生树。分支长度代表基于snv和插入缺失(indels)的相对距离,并且根据活检类型,对所述分支进行着色。通过血液样品对患者1(a)、患者2(b)和患者3(c)确立最大似然系统发生树。节点支持作为引导值给出,并且将大于50%视为强支持。
图3:患者1样品的数字pcr和sanger测序。(a)使用针对brafv600e和nrasq61k的探针的dpcr显示在所有肿瘤样品中的brafv600e突变的dna。对nrasq61k阳性的dpcr反应仅在这名患者的晚期转移瘤6中检测到。小于15%的精度值被认为是高度可重复的阳性反应。(b)代表谱图和(c)从分离自晚期转移瘤6的单黑素瘤细胞培养的26个细胞培养物的sanger测序的序列。所有26个克隆培养物都具有brafv600e和nrasq61k两个突变。
图4:在双突变黑素瘤细胞中的活力测定和perk信号转导。从患者1的晚期转移瘤6建立的抗性细胞培养物显示对不同braf抑制剂可变的反应。(a)相对于dmso处理的细胞,对测量使用不同braf抑制剂处理后的nad(p)h酶活性的一式三份mtt测定进行归一化。源自在lgx818治疗的同时发生进展的患者的抗性细胞系m121224,对lgx818具有完全抗性,但对plx4032和gsk2118436仅具有部分抗性。(b)在braf抑制剂处理后,m121224细胞中perk水平的western印迹及其定量。使用imagej测量条带的光密度并获得条形图。基于敏感性细胞系m000921,以及其他brafv600e突变的早期传代培养物的ic50选择药物浓度。(c)显示在使用0.35μmplx4032处理后的perk靶基因的相对表达的qpcr。(d)测量在使用mek抑制剂(mek162)、mek和braf抑制剂的组合(lgx818),以及单独的erk抑制剂(sch772984)的治疗后的nad(p)h酶活性的mtt测定。
图5:通过突变等位基因比例(mar)测量的亚克隆多样性。(a)患者1的原发肿瘤的突变等位基因比例的频率显示纯合、杂合的以及可能的亚克隆snv。患者1的痣和转移瘤的比较显示在原发肿瘤中提高的亚克隆频率。(b)患者2的原发肿瘤的总snv(黑线)与仅存在于患者2的原发肿瘤的第一钻孔样品(灰线)中的snv的比较。对单个钻孔样品私有的snv通常具有低mar。图下面的数据代表平均mar。
图6:源自转移性黑素瘤的单细胞克隆的双突变黑素瘤细胞的活力测定。相对于dmso处理的细胞,对测量使用mek抑制剂mek162(a)、erk抑制剂sch772984(c),或braf抑制剂gsk21184362(b)、lgx818(d)或plx4032(e)处理后的nad(p)h酶活性的一式三份mtt测定进行归一化。brafv600e和nrasq61r双突变的克隆细胞系m140307和m150423对braf抑制剂处理具有抗性,但对erk抑制剂处理敏感。
实施例
所述实施例仅说明本发明及其方面。它们不应以任何方式解释为对本发明范围的限制。
对来自三名转移性黑素瘤患者的多个样品的全外显子组进行测序,其包括不同的解剖部位,治疗和疾病进展阶段(图11a-f)。在初始诊断中,患者1具有brafv600e突变(图1a),患者2具有未知的致癌驱动(图1b),并且患者3具有活化的nrasq61r突变(图1c)。患者1接受靶向braf抑制剂(即lgx818),并且根据计算机x线断层摄影术(ct)具有部分响应(图1d)。根据pet/ct,患者2在多激酶抑制剂治疗(即帕唑帕尼)下出现进展(图1f)。患者3接受靶向mek抑制剂(即mek162),并且根据ct也出现进展(图1f)。测序结果的分析显示在肿瘤样品中预期数量的总单核苷酸变异(snv),如在此前研究中所公开的6,12。如通过具有非同义snv的基因总数所测量的,来自患者1的两个发育异常的痣具有比来自所述三明患者的任何其他肿瘤活检样品更低的蛋白编码突变负担。痣1具有133个突变基因,且痣2具有101个突变基因,而患者1的肿瘤活检样品具有平均186个突变基因。患者2和患者3在其肿瘤中分别具有196和234个突变基因。有趣的是,除了具有平均数量更少的突变基因之外,与所有其他测序的原发(1.20)和转移性黑素瘤(1.22)病灶相比,所述痣还具有降低的非同义对同义突变的比例(即0.79),表明痣与一般黑素瘤肿瘤相比蛋白编码变化的比例更小。有趣的是,还注意到每个原发肿瘤具有比患者的各个转移瘤高的私有snv数量,表明在原发肿瘤中比转移瘤升高的个体基因多样性13。例如,患者1具有原发肿瘤专有的96的个体snv,且在所有转移瘤中平均为35的个体snv。患者2具有对原发肿瘤的三个钻孔样品专有的平均为48的私有snv,以及在转移瘤中平均24的私有snv。同样,除了患者3中的一个明显异常转移瘤(即晚期1)之外,两个原发肿瘤钻孔中的每一个都具有高于转移瘤(即38)的私有snv数(即89)。因此,在我们的组群中,原发肿瘤在整体上具有比相同患者的转移瘤显著高出2-2.7倍的私有snv数(t-检验,p<.00048),但有一个异常转移瘤显示异常的个体突变数。
外显子组测序可确认已知的braf和nras突变状态,其首先在对每位患者诊断时通过sanger测序确定(图1)。此外,筛选其它已知致癌基因和肿瘤抑制因子的数据,其能够在我们的组群的黑素瘤进展中发挥作用。尽管患者2在诊断时不具有已知的致癌驱动,但确定了黑皮素受体mc1rv92m中的非同义种系突变,其已显示与获得转移性黑素瘤的风险升高显著相关14。此外,患者3具有种系突变mitfe318k,最近将其与发展黑素瘤的风险提高相关联。
为了确定这三名患者的潜在肿瘤抑制基因座的基因组丢失,使用excavator和contra算法分析外显子组数据16,17,其允许推断拷贝数变异(cnv)。可在很多染色体中检测到高cnv数值,并且一些样品显示在遍及基因组的大量丢失(图g-i)。
在所研究的组群中确定了已知常常出现在黑素瘤中的染色体不平衡(图1g-i)。患者1在晚期转移瘤3和4中在6p、7、8q和17q中的拷贝增加(图1g)。患者2在晚期转移瘤中在染色体1q、7和22中具有增加(图1h)。在患者3中,我们在染色体1q、6p和20q中发现增加(图1i)。所有患者显示在染色体6q、9p和10中,以及在一些样品中在染色体11、2和17中的至少部分丢失(图1g-i)。
此外,contra提供了关于cnv的基因特异性信息。除了来自患者1的痣中之外,在所有肿瘤样品中发现染色体9上cdkn2a基因座的一致丢失。通过qpcr证实,这些丢失在患者1和3中是纯合的,在患者2中是杂合的(数据未显示),与通过excavator和contra算法所预期的一样(图1g-i,补充表3)。此外,在患者2的所有样品(图1h)和来自患者1的大多数样品(除了早期转移1和原发肿瘤)中pten(染色体10)丢失。
将肿瘤样品分组并建立活检样品之间关系的一个方法是假设cnv一旦丢失,就不能恢复18。因此,可通过确定原发肿瘤中的特异性基因组丢失来推测肿瘤系统发生,所述特异性基因组丢失不能在由这个原发肿瘤衍生出的转移瘤中恢复。然而,所确定的患者内染色体不平衡的高可变性将导致取样的活检样品内很多不同的可能的关系(图1g-i)。例如,在患者3中,染色体10cnv可表明晚期转移瘤源自原发钻孔样品#2;然而,染色体14cnv更提示晚期谱系源自原发钻孔样品#1(图1i)。同样,在患者2中,原发钻孔样品#2具有比其他两个原发钻孔样品更少的染色体11中的丢失,这表明与晚期转移瘤较少的相似性,而在染色体3上的丢失模式则可表明原发钻孔样品#2和晚期转移瘤之间更密切的关系(图1h)。通常,患者内cnv异质性非常高,正如可以在我们对其相同原发肿瘤的多个区域进行测序的患者中所观察到的(图1h,i)。例如,在患者2的染色体11和患者3的染色体7、10、12和14中,我们仅在两个原发肿瘤钻孔样品中的一个发现丢失。cnv的异质性也可清楚见于患者1的染色体22,例如,其具有在原发肿瘤的端粒区的预期的拷贝数增加,其没有出现在任何晚期转移瘤中(图1j)。
实施例2:全外显子组系统发生分析确定肿瘤间关系和进展相关snv
为了研究在不同治疗环境中单个患者肿瘤之间的进化关系,将系统发生算法应用于来自每个患者的snv和插入/缺失调用(indelcalls)。全外显子组系统发生分析不仅允许基于其总snv、插入和缺失将肿瘤样品分组,并且还允许测定样品之间的进化关系,并且甚至还允许发现支持特异性系统发生节点的诊断特征(图2)。来自患者1和3(即分别使用braf和mek靶向抑制剂治疗)的活检样品显示具有形成单系分支的后抗性肿瘤的树,意味着所有的后抗性样品都源自唯一一个节点。通过引导支持(bootstrapsupports)(箭头)显示置信度,其反应了还解析出在该分支重点的进化枝的引导树(bootstraptrees)的百分比。接受非靶向治疗(即多受体酪氨酸激酶抑制剂帕唑帕尼)的患者2没有显示晚期肿瘤转移的这种强烈的单系支持(图2),但显示后抗性样品源自多个节点(箭头)。
在靶向治疗后,来自患者1和3的系统发生树的强单系拓扑学表明治疗抗性的机制可支持区分治疗前和治疗后进化枝的节点(图2a,c)。然而,在这些节点支持或在可以解释在患者1和3中观察到的治疗抗性的全外显子组数据中,不能确定对braf抑制剂或mek抑制剂治疗的已知和共有机制。对每个患者中的所有复发后肿瘤外显子组之间的非同义snv的交点进行研究,以发现新的潜在遗传抗性机制。在患者1中,发现tacc1l452v的体细胞非同义突变对抑制剂抗性肿瘤样品是普遍存在且专有的。尽管已经发现tacc1常常在黑素瘤肿瘤中突变,但尚未确定tacc1在治疗抗性中的作用126。由于可能有抗性机制的患者内肿瘤间异质性,寻求确定在任何治疗后样品中说明性的蛋白编码变化。在患者1中,在原发和晚期转移瘤1中检测到gnaqt96s中的非同义突变,并且在相同活检样品中的tacc1c133a也检测到。尽管这些突变位于此前显示在黑素瘤中发挥作用的基因,但它们在治疗抗性中的作用仍是未知的。同样,在其他两位患者的外显子组数据中也没有确定已知的抗性机制。
实施例3:lgx818抗性的患者内遗传异质性
由于在两个靶向治疗患者中缺少已知的共有抗性机制,进一步研究了braf抑制剂治疗的患者样品(即,患者1),这是由于文献中braf抑制剂抗性机制的信息更多9。对相同活检样品以及其他活检样品进行sanger测序,所述其他活检样品的dna在不扩增的情况下对外显子测序而言过于受限。可通过来自所有肿瘤样品的pcr扩增子的标准sanger测序确认brafv600e突变(数据未显示)。由于活化nras突变是目前确定的最常见的抗性机制,在17.8%的braf抑制剂抗性肿瘤中存在9,选择该突变用于在所有患者1样品中的nras基因座的外显子2和3的首次sanger测序。在这种情况下,确定了在复发后出现的患者1晚期转移瘤6号中的活化突变nrasq61k。相同的突变在所有其他转移样品中都不存在。此外,可以确定这个转移瘤仍具有brafv600e突变以及在患者1的其他治疗后转移瘤中专有且普遍存在的另外两个突变:tacc1l452v和c11orf30k22n(数据未显示)。没有通过sanger测序测试其他特异性突变,但源自晚期转移瘤6的原代细胞培养物(即培养物编号m121224)的后续外显子组测序,也可确认这些突变的存在。
由于全外显子组测序提供了广泛的基因组覆盖,但在具体基因座的深度有限(在我们的案例中,所有样品之间101x平均覆盖),因此难以检测具有可选基因型的癌细胞的低丰度亚克隆19,20。由于这种原因,应用数字pcr以进一步研究患者1的治疗后肿瘤中突变且抗性细胞的小型亚群体的可能性。我们的数字pcr平台是基于每次运行的20’000个同时pcr反应,其允许检测以低至5%的肿瘤细胞群体存在的基因组变体。
通过使用这种技术,我们对每个样品测量了每微升dna的brafv600e或nrasq61k突变拷贝的数量。认为具有小于15%的精密度的数值是可以接受的,其表示在测量的拷贝数附近+/-15%的置信区间。数字pcr证实brafv600e突变存在于所有的肿瘤中,但不存在于从患者血液或痣1获得的dna中(图3a)。尽管晚期转移瘤4显示低的每微升拷贝数(即35个拷贝),但精密度在可接受的范围之内(即8.59%)。然而,来自患者1的所有其他肿瘤,包括尚未进行外显子组测序的那些(即晚期转移瘤5&6)具有较高的brafv600e拷贝数和高精密度(图3a)。还通过数字pcr证实了nrasq61k突变在晚期转移瘤6中的存在,并且显示在该转移瘤中具有高拷贝数(图3a,绿色框)。数字pcr结果还显示除转移瘤6号之外,在任何其他抗性转移瘤中不存在可检测到的nrasq61k亚克隆。
实施例4:单braf抑制剂抗性,但mek和erk抑制剂敏感的黑素瘤细胞中存在两种活化mapk突变
尽管可显示mapk-活化突变brafv600e和nrasq61k都存在于来自患者1的单个后抗性肿瘤,但这些结果可以解释为存在两个分离的细胞亚群,其各自具有一个活化mapk突变,或者两个突变都存在于单个细胞中。为了区分这些可能性,通过facs分选从m121224分离单个黑素瘤细胞,并从这些单个细胞的每一个生长出新的培养物。对源自26个不同单细胞克隆的26个培养物的sanger测序可以确定brafv600e和nrasq61k突变都存在于所有23个独立衍生的群落中(图3b,c)。为了确认m121224保留了晚期转移瘤6的braf抑制剂抗性,使用lgx818和其他两种商购可得的braf抑制剂(即plx4032和gsk2128436)处理m121224,并通过mtt测定测量细胞活力(图4a)。包括brafv600e突变的黑素瘤细胞培养物(m980513)作为阳性对照,和nrasq61r突变的细胞培养为(m010817)作为braf抑制剂处理的阴性对照。m121224系仍对lgx818具有抗性,达到与brafwt细胞培养物m010817相同的程度(图4a)。同样,m121224还对plx4032和gsk2118436具有抗性,但程度低于患者对其产生抗性的lgx818抑制剂(图4a)。在lgx818和plx4032的ic50浓度下,磷酸化erk(perk)水平在m121224中显著降低(图4b)。在plx4032和lgx818的ic50浓度下,观察到m980513和m121224的三个perk靶基因的显著下调,但在对照nrasq61r细胞系中则没有。
尽管m121224双突变细胞在高浓度的lgx818药物存在下保持存活(图4a),但仍想知道两种活化mapk突变的共同存在如何影响这些细胞对其他mapk途径抑制剂的敏感性。使用标准ic50浓度的lgx818和提高浓度的mek抑制剂(mek162)二者对m121224细胞的处理,能够显示与具有单nrasq61r突变的细胞类似的活力概况(图4d)。同样,单独的mek抑制剂在降低m121224细胞活力中有效,就如同在nrasq61r突变细胞中一样(图4d)。最后,单独的特异性erk抑制剂同样消除了m121224活力,达到如在brafv600e细胞中相同的程度(图4d)。
进一步通过使用从braf抗性转移性黑素瘤分离的其他双突变克隆细胞系的试验确认对erk抑制剂处理的高敏感性(图6)。
实施例5:原发肿瘤展示最高的亚克隆性
合理假设癌症中的大部分体细胞突变影响一个等位基因但不影响其他等位基因,因此,其为杂合的。在克隆且纯的癌群体中,此类突变展示0.5的突变等位基因比例(mar),也即所有测序碱基的一半显示突变等位基因。偏离此数值可表示存在癌症亚克隆,其导致小于0.5的mar。为了研究亚克隆在我们的原发肿瘤中的存在,测定了多个钻孔样品之间的mar。此外,通过以突变数量除以总读取计数计算突变等位基因比例(mar),从而获得在样品内的潜在存在的亚克隆的量度21。对比患者1中的痣和转移瘤,在原发肿瘤中观察到较高比例的snv和低mar,其中原发肿瘤具有44%的平均mar,而所述痣和转移瘤具有50%的平均mar(图5a)。此外,由于相同的原发肿瘤被钻孔多次,mar可以是对患者2和3的原发肿瘤的所有样品。推测起来,这些钻孔样品表征了肿瘤的不同部分,并且一些突变专有地见于一个钻孔样品中,但在其他钻孔样品中没有发现(私有突变)。必须假定这些私有突变是亚克隆的,并且因此存在于较小的细胞组中。图5b中的结果清楚显示每个原发肿瘤钻孔样品的私有snv的平均mar显著低于所有snv的整体mar。来自患者2的每个钻孔样品的私有与总snv的mar在每种情况下低6%至16%之间(图5b),并且平均mar在患者3的私有snv中比总snv低9%至15%之间(图5c)。这些结果表明原发肿瘤包含最高的亚克隆多样性,其可以通过大数量的私有snv,和低突变等位基因频率来表征。然而,每个原发肿瘤钻孔样品具有不同程度的亚克隆性,表明肿瘤内在克隆多样性中的异质性。
在患者1中,观察到原发肿瘤中maf的双峰分布,一个峰在0.35且第二个峰在0.15。第一峰可能对应于克隆的杂合突变,并且表示70%的肿瘤纯度。
实施例6:实验程序
样品制备
根据伦理批准号647和800,仅在通过大学生物库计划给出患者的书面同意后使用患者材料。dna从保存在苏黎世大学医院皮肤病学研究所的生物库中的石蜡包埋组织,新鲜冷冻组织或pbmc分离。使用来自qiagen的ffpedna分离试剂盒(qiaampdnaffpetissuekit#56404)以及桑格研究院(sangerinstitute)的ultanmcdermott开发的优化方案从石蜡块分离dna。对于从非石蜡包埋样品的dna分离,我们遵循此前公开的标准dna分离方案。在得到患者同意的情况下,在死亡后不就的尸检期间收集样品。在收集后立即处理样品,以确保尽可能好的dna和rna质量。在可能的情况下,建立原代细胞培养物,如此前研究中那样28。
为了减少基质组织污染,对石蜡块钻孔,并从钻孔样品分离出dna,而不是从整块的切片分离。在dna分离之前,由经过训练的皮肤-组织病理学家评估每个肿瘤样品。检查组织质量和肿瘤含量,并标记适合dna分离的区域。在可得的情况下,从发育异常的痣、原发黑素瘤肿瘤和治疗前取得的转移瘤,以及在验尸期间取得的转移瘤进行dna测序。如果可得,对所有患者来自pbmc的种系dna进行测序,作为参照29。
文库制备和测序
通过agilent2100bioanalyzer或agilent2200tapestation测量dna质量。使用agilentsureselectv4或v5试剂盒,使用1-3µg的高质量dna制备全外显子组文库。在苏黎世大学功能基因组中心的illuminahiseq2000机器上进行测序。对于全外显子组测序,我们的测序为0.25个泳道/样品,配对末端,以及100bp读数。
全外显子组测序分析
使用改进的gatk供应线进行生物信息学分析30-32。质量控制使用“fastqc”完成33。fastq文件与参考基因组“hg19”34的比对使用“bwa”完成35。从sam至bam文件格式的转化使用“bwa”完成。通过来自“picard”的markduplicates标记pcr重复,使用realignertargetcreator(gatk)进行插入缺失周围的局部重比对,使用indelrealigner(gatk)进行重比对,使用fixmateinformation(picard)修复匹配信息,使用baserecalibrator(gatk)和printreads(gatk)进行碱基质量得分重新校准。使用unifiedgenotyper(gatk)完成变量调用。对于vcf文件的注释,我们使用annovar37。此外,我们还使用了samtools38和bedtools39。对于数据解释,使用了microsoftaccess、microsoftexcel、venny40、conset41和igv42,43。
对所有样品计算突变等位基因频率,以获得非肿瘤组织污染程度的影响。大多数样品显示0.4至0.5的突变等位基因频率,其对应于接近100%的肿瘤材料(数据未显示)。
对于拷贝数分析,我们使用了excavator17和contra16,使用circos将使用excavator的分析结果可视化44。
根据读计数标准过滤snv:碱基必须具有至少4的突变读数和至少10的总读数,如果小于10的突变读数,则其至少一半必然是突变的。同样,将phred级别质量得分<50的所有snv从进一步分析中排除。如果来自相同患者的位过滤的血液样品对这个位置没有显示任何突变读数,则snv被称作体细胞的。
对每个调取的snv,通过以突变读数计数除以总读数计数计算突变等位基因比例(mar)。计算这些比例的频率,并在excel中使用移动平均法(周期:3)绘制趋势线。为了减少假阳性snv的数量,在私有snv上应用更严格的过滤。将质量阈值提高至100的phred得分,并且snv需要具有至少10的总读数。排除具有多于8snv的基因。
dpcr
使用abgeneamppcr系统9700(appliedbiosystemscarlsbad,ca,usa)进行数字pcr,并且使用15µl提供的主混合液(abquantstudio3d)和来自microsynth(balgach,switzerland)的等量(0.6µm)的引物。
braf正向:5`ctactgttttcctttacttactacacctcaga
反向:5`atccagacaactgttcaaactgat
nras正向:5`ggtgaaacctgtttgttggacat
反向:5`tgtattggtctctcatggcactgt
此外,我们使用了来自lifetechnologies(carlsbad,ca,usa)的探针:
brafv600e:6-vic-tagctacagagaaatc-mgb
nrasq61k:6-fam-cagctggaaaagaa-mgb
将dna稀释至4µm的终浓度;取决于靶序列的预期频率,dna浓度在0.3ng/µl至6.6ng/µl变化。芯片加载和热循环条件参照lifetechnologies说明书。使用quantstudio3d进行荧光测量,并通过quantstudio3danalysissuite软件处理输出值。对荧光数值进行泊松校正,并计算每µl拷贝数。将显示高于15%的精准度的每个样品归类为对特异突变阴性。
sanger测序
在dna扩增后,将12ng的各pcr产物,5xterminator测序缓冲液(appliedbiosystems),1.5µm引物(microsynth):
braf正向:5`ctaagaggaaagatgaagtactatg
反向:5`ctagtaactcagcagcatctcag
nras正向:5`gataggcagaaatgggcttga
反向:5`atcatcctttcagagaaaataatgc
和2µl的bigdye现成反应混合物(appliedbiosystems)添加至10µl反应混合物。循环条件按如下进行:在labcycler(sensoquest,göttingen,germany)中,96°c60秒,随后为96°c10秒,50°c5秒和60°c240秒的16个循环。使用bigdyexterminator纯化试剂盒(appliedbiosystems),根据制造商的手册纯化样品。使用3500geneticanalyzer(appliedbiosystems)进行随后的sanger测序。使用variantreporter软件(lifetechnologies)进行分析,其中超过25%的阈值的序列中的每个突变都归类为阳性。
细胞分选
为了进行黑素瘤细胞的单细胞分选,将来自汇合的t75细胞培养瓶的细胞沉淀,并重悬在100µlfacs缓冲液(pbs中,1%fbs,5mmedtaph8,0.01%nan3/ddh2o)中。将细胞与以下光敏抗体一起在4°c温育20分钟:抗人mcsp-fitc(miltenyibiotec130-098-794,bergischgladbachgermany),以1:20稀释在facs缓冲液中;抗人成纤维细胞/上皮-pe(abin319868,aachengermany),以1:200稀释在facs缓冲液中。在清洗后,将细胞重悬在200µlfacs缓冲液中,并使用ariaiib(bdbiosciences,franklinlakes,newjersey,usa)分选。
从pbmc分离黑素瘤细胞
根据制造商的说明,使用来自miltenyibiotec(bergischgladbach,germany)的cd56+cd16+nk细胞分离试剂盒,用1x107个pbmc分离黑素瘤细胞。与手册的一个偏差在最后一步中,其为对nk细胞的阳性选择,但流通物包含黑色素细胞;其他非nk免疫细胞在第一步中被耗尽。在收集包含所有非免疫细胞的流通物后,使细胞以1500rpm5分钟沉淀,并且随后如本文报道的非石蜡样品那样进行dna分离。
系统发生分析:
使用posix-threads版本的raxmlv8.0.19(7),构建最大简约性,贝叶斯和最大似然(ml)系统发生。将负责使用γ分布和四个速率分类的位点间速率异质性的确定偏差校正和一般时间可逆(gtr)替代模型(asc_gtrgamma模型)用于计算最优树的计算。使用100个非参数自举伪复制(nonparametricbootstrappseudoreplicates)来评估节点支持,通过引导树过滤最优ml树。因此,节点支持值表示包含给定节间分支的引导树的百分比例。
对给定进化枝为诊断性的变体定义为对于该位置仅存在于该进化枝,而不存在于他处。来自所讨论的节点发出的所有叶必须共有变体,并且所有其他叶必须包含对于待诊断的变体的不同特征。因此,诊断性变体也可称作衍征(apomorphy)。
细胞培养
根据伦理批准号647和800,通过大学生物库计划获得知情同意后,从患者的皮肤黑素瘤和黑素瘤转移的活检样品获得细胞培养物。将肿瘤材料切成小块,并使用rpmi1640(invitrogen(carlsbad,ca,usa))中的2.4u/ml分散酶(roche,basel,switzerland),在37°c降解3小时。随后,将所述材料离心(1500rpm/5min),并弃去上清。此后,将沉淀溶解在tris-缓冲盐水(ph7.4)中的0.005m二水合氯化钙和62.5u/ml胶原酶(sigma,st.louis,mo,usa)中,并在37°c温育2小时。随后,将所述材料离心(1500rpm/5min),并弃去上清。添加终止溶液(h2o中的0.05mtrisbase,0.15mnacl和0.01medta,终ph7.4),进行10分钟。此后,使用rpmi1640将沉淀清洗2次,并且最后,将细胞在补充了5mml-谷氨酰胺(biochrom,berlin,germany),1mm丙酮酸钠(gibco,carlsbad,ca,usa)和10%fcs(gibco(carlsbad,ca,usa))的rpmi1640中,在37°c和5%co2气氛中培养。在继代数次后,通过免疫组织化学确认黑素瘤培养物,并评估细胞的突变状态。
细胞活力测定
评估细胞培养物m980513(brafv600e,nraswt)、m000921(brafv600e,nraswt)、m010817(brafwt,nrasq61r)和m121224(brafv600e,nrasq61k)对不同小分子抑制剂的细胞敏感性。接种1x10^4个细胞,并使用不同浓度的braf抑制剂(plx4032,lgx818或gsk2118436)、mek抑制剂(mek162)、erk抑制剂(sch772984),或braf和mek抑制剂的组合(lgx818+mek162)处理72小时。使用dmso处理作为对照。72小时后,除去培养基,并添加补充了10%fcs和8%mtt试剂(sigma,pbs中5mg/ml)的新鲜rpmi1640,并将细胞在37°c温育。1小时后,除去rpmi1640和mtt试剂,并添加10%sds(sigma)和95%异丙醇/5%甲酸(sigma)(比例1:1)。在37°c温育5分钟后,使用微板读数仪测量595nm的吸光度(参照620nm)。
western印迹
通过使用冰冷pbs清洗细胞两次,并随后在ripa缓冲液(20mmtris-hcl(ph7.5),1%tritonx-100(sigma),137mmnacl,10%甘油和蛋白酶抑制剂(roche))中裂解,来收集总蛋白。根据制造商的方案,使用bio-raddc蛋白测定(bio-rad,hercules,ca,usa)测量蛋白的浓度。使用sds-page分离蛋白,之后将其转移到硝酸纤维素膜上。先后使用兔抗-perk抗体(cellsignaling,产品号#4376s)和兔抗-gapdh抗体(abcam,cambridge,uk,产品号ab9385),随后使用辣根过氧化物酶-缀合的山羊抗兔igg(santacruz,产品号sc-2030)探测膜。使用化学发光(ecl,gehealthcare,chalfontst.giles,uk)检测结合的抗体。此后,使用imagej软件(imagej.nih.gov/ij/)测量条带强度,并针对对应的gapdh条带强度矫正perk条带强度。
qpcr分析
使用trizol(lifetechnologies)从细胞培养物提取总rna,并且此后使用反转录系统(a3500,promega,madison,wi,usa)将1µg的rna转录成cdna。对于q-pcr,使用viia7(lifetechnologies),并且反应混合物由5µlsybrgreen(roche),3.5µlh2o,0.5µl正向+反向引物(10µm)(microsynth)和1µl的cdna(50ng)组成。
循环条件为:95℃10分钟,随后为95℃10秒和58℃30秒的40个循环,以95℃15秒,60℃1分钟和95℃15秒结束。
使用δδct方法计算perk靶基因dusp6、spry2和egr1(pmid19251651)的基因表达差别。使用gapdh作为管家基因。
引物序列
gapdh正向:gaaggtgaagttcggagtc
反向:gaagatggtgatgggatttc
dusp6正向:gaaatggcgatcagcaagacg
反向:cgacgactcgtatagctcctg
spry2正向:atcagatcagagccatccgaa
反向:tggagtctctcgtgtttgtgc
egr1正向:ggtcagtggcctagtgagc
反向:tgctgtcgttggatggcac。
申请全文引用的文献
1.bollagg,hirthp,tsaij,etal.clinicalefficacyofarafinhibitorneedsbroadtargetblockadeinbraf-mutantmelanoma.nature.2010;467(7315):596-599.
2.chapmanpb,hauschilda,robertc,etal.improvedsurvivalwithvemurafenibinmelanomawithbrafv600emutation.nengljmed.2011;364(26):2507-2516.
3.flahertykt,puzanovi,kimkb,etal.inhibitionofmutated,activatedbrafinmetastaticmelanoma.nengljmed.2010;363(9):809-819.
4.aplinae,kaplanfm,shaoy.mechanismsofresistancetorafinhibitorsinmelanoma.jinvestdermatol.2011;131(9):1817-1820.
5.maleycc,galipeaupc,finleyjc,etal.geneticclonaldiversitypredictsprogressiontoesophagealadenocarcinoma.natgenet.2006;38(4):468-473.
6.krauthammerm,kongy,habh,etal.exomesequencingidentifiesrecurrentsomaticrac1mutationsinmelanoma.natgenet.2012;44(9):1006-1014.
7.takatam,moritar,takeharak.clonalheterogeneityinsporadicmelanomasasrevealedbyloss-of-heterozygosityanalysis.intjcancer.2000;85(4):492-497.
8.wilmottjs,tembev,howlejr,etal.intratumoralmolecularheterogeneityinabraf-mutant,brafinhibitor-resistantmelanoma:acaseillustratingthechallengesforpersonalizedmedicine.molcancerther.2012;11(12):2704-2708.
9.vanallenem,waglen,suckera,etal.thegeneticlandscapeofclinicalresistancetorafinhibitioninmetastaticmelanoma.cancerdiscov.2014;4(1):94-109.
10.nazarianr,shih,wangq,etal.melanomasacquireresistancetob-raf(v600e)inhibitionbyrtkorn-rasupregulation.nature.2010;468(7326):973-977.
11.shih,hugow,kongx,etal.acquiredresistanceandclonalevolutioninmelanomaduringbrafinhibitortherapy.cancerdiscov.2014;4(1):80-93.
12.hodise,watsonir,kryukovgv,etal.alandscapeofdrivermutationsinmelanoma.cell.2012;150(2):251-263.
13.nekrutenkoa,makovakd,liwh.thek(a)/k(s)ratiotestforassessingtheprotein-codingpotentialofgenomicregions:anempiricalandsimulationstudy.genomeres.2002;12(1):198-202.
14.fernandezl,milner,bravoj,etal.mc1r:threenovelvariantsidentifiedinamalignantmelanomaassociationstudyinthespanishpopulation.carcinogenesis.2007;28(8):1659-1664.
15.berwickm,macarthurj,orlowi,etal.mitfe318k'seffectonmelanomariskindependentof,butmodifiedby,otherriskfactors.pigmentcellmelanomares.2014;27(3):485-488.
16.lij,lupatr,amarasinghekc,etal.contra:copynumberanalysisfortargetedresequencing.bioinformatics.2012;28(10):1307-1313.
17.magia,tattinil,cifolai,etal.excavator:detectingcopynumbervariantsfromwhole-exomesequencingdata.genomebiol.2013;14(10):r120.
18.schwarzrf,trinha,siposb,brentonjd,goldmann,markowetzf.phylogeneticquantificationofintra-tumourheterogeneity.ploscomputbiol.2014;10(4):e1003535.
19.flahertyp,natsoulisg,muralidharano,etal.ultrasensitivedetectionofraremutationsusingnext-generationtargetedresequencing.nucleicacidsres.2012;40(1):e2.
20.gerstungm,beiselc,rechsteinerm,etal.reliabledetectionofsubclonalsingle-nucleotidevariantsintumourcellpopulations.natcommun.2012;3:811.
21.nik-zainals,vanloop,wedgedc,etal.thelifehistoryof21breastcancers.cell.2012;149(5):994-1007.
22.guoj,sil,kongy,etal.phaseii,open-label,single-armtrialofimatinibmesylateinpatientswithmetastaticmelanomaharboringc-kitmutationoramplification.jclinoncol.2011;29(21):2904-2909.
23.asciertopa,schadendorfd,berkingc,etal.mek162forpatientswithadvancedmelanomaharbouringnrasorval600brafmutations:anon-randomised,open-labelphase2study.lancetoncol.2013;14(3):249-256.
24.emerycm,vijayendrankg,zipsermc,etal.mek1mutationsconferresistancetomekandb-rafinhibition.procnatlacadsciusa.2009;106(48):20411-20416.
25.waglen,emeryc,bergermf,etal.dissectingtherapeuticresistancetorafinhibitioninmelanomabytumorgenomicprofiling.jclinoncol.2011;29(22):3085-3096.
26.nissanmh,pratilasca,jonesam,etal.lossofnf1incutaneousmelanomaisassociatedwithrasactivationandmekdependence.cancerres.2014;74(8):2340-2350.
27.poulikakospi,persaudy,janakiramanm,etal.rafinhibitorresistanceismediatedbydimerizationofaberrantlysplicedbraf(v600e).nature.2011;480(7377):387-390.
28.widmerds,chengpf,eichhoffom,etal.systematicclassificationofmelanomacellsbyphenotype-specificgeneexpressionmapping.pigmentcellmelanomares.2012.
29.böyuma.isolationofmononuclearcellsandgranulocytesfromhumanblood.isolationofmonuclearcellsbyonecentrifugation,andofgranulocytesbycombiningcentrifugationandsedimentationat1g.scandjclinlabinvestsuppl.1968;97:77-89.
30.mckennaa,hannam,bankse,etal.thegenomeanalysistoolkit:amapreduceframeworkforanalyzingnext-generationdnasequencingdata.genomeres.2010;20(9):1297-1303.
31.vanderauweraga.fromfastqdatatohigh-confidencevariantcalls:thegenomeanalysistoolkitbestpracticespipeline.in:g.a.c,ed.currentprotocolsinbioinformatics:wileyonlinelibrary;2013.
32.depristoma,bankse,poplinr,etal.aframeworkforvariationdiscoveryandgenotypingusingnext-generationdnasequencingdata.natgenet.2011;43(5):491-498.
33.andrewss.fastqcaqualitycontroltoolforhighthroughputsequencedata.http://www.bioinformatics.babraham.ac.uk/projects/fastqc/.
34.landeres,lintonlm,birrenb,etal.initialsequencingandanalysisofthehumangenome.nature.2001;409(6822):860-921.
35.lih,durbinr.fastandaccurateshortreadalignmentwithburrows-wheelertransform.bioinformatics.2009;25(14):1754-1760.
36.picard.http://picard.sourceforge.net/.
37.wangk,lim,hakonarsonh.annovar:functionalannotationofgeneticvariantsfromhigh-throughputsequencingdata.nucleicacidsres.2010;38(16):e164.
38.lih,handsakerb,wysokera,etal.thesequencealignment/mapformatandsamtools.bioinformatics.2009;25(16):2078-2079.
39.quinlanar,hallim.bedtools:aflexiblesuiteofutilitiesforcomparinggenomicfeatures.bioinformatics.2010;26(6):841-842.
40.oliverosjc.venny.aninteractivetoolforcomparinglistswithvenndiagrams.2007;http://bioinfogp.cnb.csic.es/tools/venny/index.html.
41.kimb,leeb,seoj.visualizingsetconcordancewithpermutationmatricesandfandiagrams.interactcomput.2007;19(5):630-643.
42.thorvaldsdóttirh,robinsonjt,mesirovjp.integrativegenomicsviewer(igv):high-performancegenomicsdatavisualizationandexploration.briefbioinform.2013;14(2):178-192.
43.robinsonjt,thorvaldsdóttirh,wincklerw,etal.integrativegenomicsviewer.natbiotechnol.2011;29(1):24-26.
44.krzywinskim,scheinj,biroli,etal.circos:aninformationaestheticforcomparativegenomics.genomeres.2009;19(9):1639-1645.
45.sensi,m.,nicolini,g.,petti,c.,etal.mutuallyexclusivenrasq61randbrafv600emutationsatthesingle-celllevelinthesamehumanmelanoma.oncogene,2006,25(24),3357-3364。
- 还没有人留言评论。精彩留言会获得点赞!