高转化率和选择性ODH方法与流程
本发明涉及低级链烷烃以高转化率和高选择性方法氧化脱氢。至今,这项技术已着重于必须在反应混合物中低于氧化燃烧下限进行氧化脱氢反应。由于反应“缺氧”,单程转化率倾向于低。然而,另一方面,可高于氧化燃烧上限操作。此方法对于进料流和催化剂二者具有很短停留时间,并提供单程高转化率和高选择性方法。优选反应在用于流化床催化剂裂化器的设计装置中进行。
技术背景
链烷烃氧化脱氢成烯烃(odh)这一概念从至少二十世纪六十年代后期流行。在二十世纪六十年代之前,链烷烃蒸汽裂化是一项沿用已久的技术,并且很好地在工业上实施。odh的认知益处是较低操作温度,这进而减少温室气体排放。odh过程的不利方面是可能会分解(decomp)。工业规模设备昂贵,并且公司避开可导致分解的方法。因此,odh技术有过一段受阻的困难时期。
在二十世纪六十年代后期颁发的转让给petro-texchemicalcorporation公司的一些美国专利公开在蒸汽裂化器中用不同的铁酸盐从链烷烃制备烯烃。这些专利包括以woskow等人名义的美国专利3,420,911和3,420,912。这些专利教导引入铁酸盐,例如,铁酸锌、铁酸镉和铁酸锰(即,与氧化铁的混合氧化物)。铁酸盐在约250℃至约750℃温度在小于100psi(689.476kpa)的压力引入脱氢区域小于2秒时间,一般0.005至0.9秒。反应似乎在蒸汽存在下进行,蒸汽可倾向于以“错误的”方向使平衡移动。另外,反应并非在催化剂存在下进行。
英国专利1,213,181,似乎部分对应以上petro-tex专利,公开可在氧化脱氢过程中使用铁酸镍。反应条件可与以上提到的petro-tex专利相比。
2005年5月10日颁予liu、转让给symyxtechnologies,inc.公司的美国专利6,891,075教导一种用于链烷烃(烷烃,例如乙烷)氧化脱氢的催化剂。气体原料包含至少烷烃和氧,但也可包括稀释剂(例如,氩、氮等)或其它组分(例如,水或二氧化碳)。脱氢催化剂包含至少约2%重量nio和宽范围其它元素,优选nb、ta和co。当nio存在于催化剂时,它似乎不是烷烃(乙烷)氧化脱氢的氧源。
2003年2月18日颁予ozkan等、转让给俄亥俄州立大学(ohiostateuniversity)的美国专利6,521,808教导用于乙烷氧化脱氢成乙烯的溶胶-凝胶负载型催化剂。催化剂似乎为混合二氧化硅/氧化钛载体上的混合金属系统,例如ni-co-mo、v-nb-mo,可掺有少量li、na、k、rb和cs。同样,催化剂不提供氧化脱氢所用的氧,而是在进料中包含气态氧。
1984年5月22日颁予eastman等、转让给phillipspetroleumcompany公司的美国专利4,450,313教导一种组成为li2o-tio2的催化剂,其特征为不超过10%的低乙烷转化率,然而有到乙烯的相当高选择性(92%)。这种催化剂的主要缺陷是氧化脱氢过程的高温,接近或高于650℃。
1986年6月24日颁布的转让给unioncarbidecorp.公司的美国专利4,596,787a公开用于乙烷低温氧化脱氢成乙烯的负载型催化剂的制备。用于乙烷低温气相氧化脱氢成乙烯的负载型催化剂通过以下步骤制备:(a)制备具有金属化合物可溶部分和不可溶部分的前体溶液,(b)分离可溶部分,(c)用可溶部分浸渍催化剂载体,和(d)活化经浸渍载体,以得到催化剂。经煅烧催化剂具有组成moavbnbcsbdxe。没有x或x为li、sc、na、be、mg、ca、sr、ba、ti、zr、hf、y、ta、cr、fe、co、ni、ce、la、zn、cd、hg、al、tl、pb、as、bi、te、u、mn和/或w;a为0.5-0.9;b为0.1-0.4;c为0.001-0.2;d为0.001-0.1;在x为元素时,e为0.001-0.1。该专利未能教导或提出催化剂和载体共研细。
乙烷用包含钼、钒、铌和锑的经煅烧氧化物催化剂低温氧化脱氢成乙烯的另一个实例描述于1985年6月18日颁发的美国专利4,524,236a和1981年2月10日颁发的4,250,346a,两专利均转让给unioncarbidecorp.公司。该经煅烧催化剂包含氧化物形式的moavbnbcsbdxe。催化剂从各金属的可溶化合物和/或络合物的溶液和/或化合物制备。经干燥催化剂通过在空气或氧中在220-550℃加热来煅烧。可使催化剂前体溶液负载于载体上,例如二氧化硅、氧化铝、碳化硅、氧化锆、氧化钛或这些的混合物。对于50%乙烷转化率,到乙烯的选择性可大于65%。
2003年9月23日颁予bharadwaj等的美国专利6,624,116和2003年5月20日颁予bharadwaj等的美国专利6,566,573,两专利均转让给dowglobaltechnologiesinc.公司,公开已在自热范围在т>750℃试验的pt-sn-sb-cu-ag整体式系统,起始气体混合物包含氢(h2:o2=2:1,ghsv=180000h-1)。催化剂组成不同于本发明催化剂的组成,本发明不预期在进料中使用分子氢。
1985年6月18日颁予mccain、转让给unioncarbidecorporation公司的美国专利4,524,236和1990年2月6日颁予manyik等、转让给unioncarbidechemicalsandplasticscompanyinc.公司的美国专利4,899,003公开v-mo-nb-sb的混合金属氧化物催化剂。在375-400℃,乙烷转化率以接近71-73%的选择性达到70%。然而,这些参数只在小于900h-1(即,720h-1)的很低气体时空间速度达到。
2008年1月15日颁予lopez-nieto等、转让给consejosuperiordeinvestigacionescientificasanduniversidadpolitecnicadevalencia的美国专利7,319,179公开mo-v-te-nb-o氧化物催化剂,该催化剂在360-400℃提供50-70%乙烷转化率和高达95%到乙烯的选择性(在38%转化率)。催化剂具有经验式motehvinbjakox,其中a为第五改性元素。该催化剂为经煅烧混合氧化物(至少mo、te、v和nb的氧化物),任选负载于:(i)二氧化硅、氧化铝和/或氧化钛,优选全部负载型催化剂的20-70%重量的二氧化硅;或(ii)碳化硅上。负载型催化剂通过从溶液沉淀、干燥沉淀然后煅烧的常规方法制备。
制备mo-te-v-nb组合物描述于2005年6月30日公布的wo2005058498a1(对应于已公布美国专利申请2007149390a1)。制备催化剂包括,通过惰性陶瓷载体与至少一种包含mo、v、te和nb离子物类的溶液组合制备浆料,干燥浆料以得到颗粒产物,在含氧气氛在150-350℃预煅烧经干燥产物,并在惰性气氛下在350-750℃煅烧经干燥产物。制备的催化剂显示在氧化反应中可与非负载型催化剂相比的活性和选择性。
在2006年12月7日公布的转让给celaneseint.corp.公司的wo2006130288a1公开一种从包含乙烷和氧的气体进料制备乙烯的方法,所述方法包括使进料与含钒、钼、钽和碲的混合氧化物催化剂在反应器中接触,以生成乙烯的流出物。催化剂具有50-80%的乙烯选择性,从而使乙烷以高选择性氧化产生乙烯和乙酸。催化剂具有式mo1v0.3ta0.1te0.3oz。催化剂任选负载于载体上,载体选自多孔二氧化硅、灼烧的二氧化硅、硅藻土、硅胶、多孔和无孔氧化铝、二氧化钛、二氧化锆、二氧化钍、氧化镧、氧化镁、氧化钙、氧化钡、氧化锡、氧化铈、氧化锌、氧化硼、氮化硼、碳化硼、磷酸硼、磷酸锆、硅酸铝、氮化硅、碳化硅和玻璃、碳、碳纤维、活性炭、金属氧化物或金属网络和相应的整料,或者包封于材料中,优选二氧化硅(sio2)、五氧化二磷(p2o5)、氧化镁(mgo)、三氧化铬(cr2o3)、氧化钛(tio2)、氧化锆(zro2)或氧化铝(al2o3)。然而,制备负载型组合物的方法包括湿化学步骤(溶液浸入固体载体,然后将材料干燥和煅烧)。
1993年4月13日颁予minet等、转让给medalertincorporated公司的美国专利5,202,517教导用于乙烷常规脱氢成乙烯的陶瓷管。“管”为一种陶瓷膜,乙烷在管内流动,氢扩散出管,以改善反应动力学。反应性陶瓷在1.5至2mm厚载体上为5微米厚。
2004年11月16日颁予adris等、转让给sabic的美国专利6,818,189教导在通道桥接塔(passagebridgingcolumns)9和10中的一种方法,其中陶瓷粒料填充在管式反应器周围,且不同的反应剂围绕管外和管内流动。专利涉及乙烷氧化脱氢成乙烯。
有相当多技术用+1氧化态的银或铜离子分离乙烯和乙烷。参见美国专利6,518,476第5栏10-15行,和第16栏12行-第17栏57行。novachemicals公司也公开用离子液体(二硫纶,在ca2,415,064中(现在已放弃))从非烯烃分离烯烃。也参见颁予exxon的美国专利6,120,692;2003年2月11日颁予unioncarbide的美国专利6,518,476,第16栏和第17栏;1984年9月29日公开的jp59172428的摘要;和1984年9月29日公开的jp59172427的摘要。
2011年9月13日颁予kuznicki等、转让给阿尔伯塔大学(theuniversityofalberta)的美国专利8,107,825包含用ets-10从乙烯分离乙烷和吸附方法的现有技术的很好概括。
2008年8月12日颁予lucy等、转让给bpchemicalslimited公司的美国专利7,411,107公开一种用于从乙烷转化成乙烯和乙酸的氧化脱氢过程分离乙酸的方法。该方法用金属盐(例如,cu或ag)的可逆络合物分离乙烯(第8栏)。专利然后公开,可通过蒸馏从液体分离乙酸(第13栏35至40行)。
以novachemicals(international)s.a.名义的美国专利申请20110245571教导乙烷在与提供氧到反应器的再生氧化物床接触的流化床中氧化脱氢。在此方法中,“游离”氧不直接与原料混合,从而减小“分解”的可能性。
1975年9月9日颁予lo等、转让给elpasoproductscompany公司的美国专利3,904,703教导一种分区或分层的氧化反应器,其中在氧化脱氢的区域后有“氧化区域”,在脱氢区域后使氢氧化成水。在氧化区域后有吸附床,以在它们进入随后脱氢区域之前从反应剂除水。这是要减小水对下游脱氢催化剂的影响。
2010年10月7日以arnold等名义公布、转让给lummus的美国专利申请2010/0256432在第86-94段中公开从产物流去除残余氧的方法。可向产物流加入可燃物,例如氢或烃,以除去残余氧。专利涉及催化剂,但未公开其组成。如上提到,然后可有必要处理产物流,以除去水。
2004年1月15日以wang等名义公布、转让给conocophillipscompany公司的美国专利申请2004/0010174(现已放弃)公开用循环流化床(cfb)反应器(设计上与fcc反应器相似)进行氧化脱氢。该公开在第40段教导催化剂用于将氧作为晶格氧或作为吸附氧带入反应器。该公开与将空气或氧加到进料流的教导相反。
2013年8月27日颁予arnold等、转让给lummustechnologyinc.公司的美国专利8,519,210教导,进料中的氧浓度可在下方有余量的情况下限于燃烧的最低氧,一般通过包括蒸汽或惰性气体以稀释进料到低于燃烧限。
本发明寻求提供低级链烷烃(烷烃,优选正烷烃)氧化脱氢产生α-烯烃的单程方法。
发明公开内容
在一个实施方案中,本发明提供一种或多种选自乙烷和丙烷及其混合物的烷烃在负载型催化剂存在下氧化脱氢的方法,所述负载型催化剂选自:
i)下式的催化剂:
vxmoynbztemmenop
其中me为选自ta、ti、w、hf、zr、sb及其混合物的金属;并且
x为0.1至3,其条件为在me不存在时,x大于0.5;
y为0.5至1.5;
z为0.001至3;
m为0.001至5;
n为0至2;并且
p为满足混合氧化物催化剂价态的数;
ii)下式的催化剂:
moavbnbcteeod
其中:
a为0.75至1.25,优选0.90至1.10;
b为0.1至0.5,优选0.25至0.4;
c为0.1至0.5,优选0.1至0.35;
e为0.1至0.35,优选0.1至0.3;并且
d为满足金属氧化物载体上混合氧化物催化剂价态的数;
所述方法包括:
a)在250℃至450℃温度,3.447至689.47kpa压力(0.5至100psi),优选103.4至344.73kpa(15至50psi),和所述一种或多种烷烃在所述反应器中0.002至10秒停留时间,使所述一种或多种烷烃和氧通过包含所述催化剂的流化床的氧化脱氢反应器,并还原所述催化剂,所述催化剂具有在脱氢反应器中小于30秒平均停留时间;
b)将所述经还原催化剂送到再生反应器,并使空气流任选连同添加的氮在250℃至400℃温度和3.447至689.47kpa(0.5至100psi)压力[优选103.4至344.73kpa(15至50psi)]通过所述床,以氧化所述催化剂;并且
c)使所述经氧化催化剂通回到所述氧化脱氢反应器,其中到所述反应器的进料中氧的量高于所述进料的燃烧上限。烷烃到烯烃的单程转化率不小于50%,烷烃转化成烯烃的选择性不小于0.9。
在另一个实施方案中,方法包括使产物流通过一个或多个氧清除反应器。优选反应器区域平行操作,且一个正经氧化,而另一个正经还原,以降低催化剂中金属的氧化态。
在一些实施方案中,氧清除反应器使用氧化脱氢反应器中所用相同的催化剂。
在另一个实施方案中,氧化脱氢反应器包括升管,再生反应器为单独的流化床反应器,所述再生反应器与所述升管连接,以使经氧化催化剂流回到所述升管(例如,cfb[循环流化床]型反应器)。
在另一个实施方案中,所述升管顶部包括分布器系统,以改善反应器中温度控制[以使烷烃进料燃烧最大限度地减少并且]以保持反应器的总选择性高于90%。
另一个实施方案包括使一个或多个低温蒸汽和雾化水通到进入所述升管的所述催化剂流中,以冷却催化剂,控制氧化脱氢反应器的热平衡。
在另一个实施方案中,在所述氧化脱氢反应器和所述再生反应器之间有降管,以使经还原催化剂从所述氧化脱氢反应器流到所述再生反应器。
另一个实施方案包括通入低温蒸汽[相对于催化剂流逆流通过所述降管],以汽提夹带的烷烃进料和产物。
另一个实施方案包括使空气或空气和氮的混合物以一定量通过再生反应器,以便从空气或空气和氮的混合物基本提取出氧,并产生包含不小于85%体积氮的气体产物流。
另一个实施方案包括使氧减少的流出物流的一部分从再生器反应器再循环,任选使其冷却,并使其再循环回到再生器反应器。
在另一个实施方案中,将co促进剂加到再生器反应器。
另一个实施方案包括使来自氧化脱氢反应器的所述烯烃产物与来自氧化脱氢单元的产物流中的水分离。
另一个实施方案包括使来自流出物流的未用氮从催化剂再生反应器通到用氮作为部分原料的现场集成装置操作。
在另一个实施方案中,用两个或更多个固定床反应器作为清除器,固定床反应器具有管和阀,以便到流化床氧化脱氢反应器的进料通过一个或多个具有氧化脱氢催化剂的固定床反应器,所述进料被氧化使催化剂耗减氧;并使产物流通过一个或多个具有耗减氧的氧化脱氢催化剂的固定床反应器,通过反应,并使到反应器的产物流切换到耗减氧的反应器,和使进料流切换到富氧反应器,从产物去除残余氧。
在另一个实施方案中,现场集成单元操作选自氨装置和丙烯腈装置、脲装置和硝酸铵装置。
在另一个实施方案中,催化剂在氧化脱氢反应器中的停留时间小于30秒(优选小于10秒,更理想小于5秒)。
在另一个实施方案中,催化剂在再生反应器中的停留时间小于3分钟。
在另一个实施方案中,催化剂在再生器中的停留时间与催化剂在氧化脱氢催化剂中的停留时间之比不小于3。
在另一个实施方案中,来自氧化脱氢反应器的产物流和来自再生器反应器的至少部分流出物流通过单独的蒸汽发生器以回收热。
在另一个实施方案中,使来自氧化脱氢反应器的产物流冷却,并通过塔,以从烯烃分离燃烧产物。
在另一个实施方案中,使来自氧化脱氢反应器的产物流冷却,并通过胺单元,以去除co2。
在另一个实施方案中,载体选自二氧化硅、熔融二氧化硅、氧化铝、二氧化钛、二氧化锆、二氧化钍、氧化镧、氧化镁、氧化钙、氧化钡、氧化锡、二氧化铈、氧化锌、氧化硼、氧化钇。
在另一个实施方案中,烷烃为乙烷。
在另一个实施方案中,催化剂具有下式:
moavbnbcteeod
其中:
a为0.90至1.10;
b为0.25至0.4;
c为0.1至0.3;
e为0.1至0.3;并且
d为满足金属氧化物载体上混合氧化物催化剂价态的数。
在另一个实施方案中,在氧化脱氢反应器中,到乙烯的转化率大于60%。
在另一个实施方案中,在氧化脱氢反应器中,到乙烯的选择性大于75%。
附图简述
图1为根据本发明可用的cfb反应器的示意图。
图2a和2b显示根据本发明在催化剂存在下在355℃温度经长达60秒时间,包含乙烯和25%mol氧的进料流的转化率和选择性。
图3a和3b显示根据本发明在催化剂存在下在355℃温度经长达60秒时间,包含乙烯和25%mol氧的进料流的转化率和选择性。
图4为显示在398℃在mo-v-te-nb-ox催化剂上气流切换[空气到气体混合物]后乙烷和o2转化率(a)和乙烯生成选择性(b)的时间依赖性的绘图。[2400h-1]虚线对应于平衡值。
图5为显示在398℃在mo-v-te-nb-ox催化剂上气流切换[空气到气体混合物]后乙烷和o2转化率(a)和乙烯生成选择性(b)的时间依赖性的绘图。[600h-1]虚线对应于平衡值。
图6为显示在398℃在不同流速mo-v-te-nb-ox催化剂上气流切换[空气到气体混合物]后乙烯生成选择性的时间依赖性的绘图。
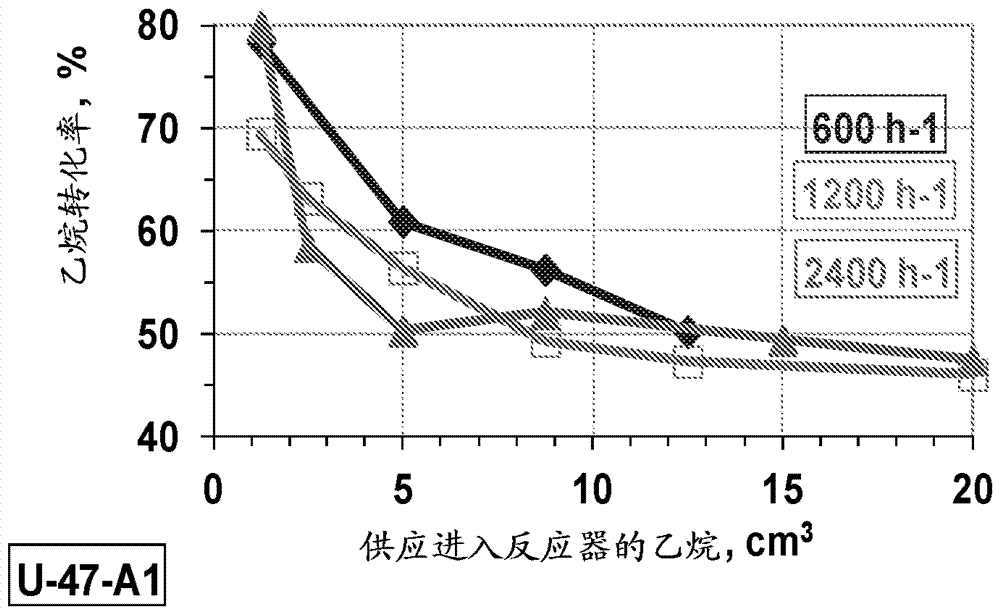
图7为显示在398℃在mo-v-te-nb-ox催化剂上气流切换[空气到气体混合物]后乙烷转化率对供应进入反应器的乙烷的量的依赖性的绘图。
图8为显示在355℃在mo-v-te-nb-ox催化剂上气流切换[o2到c2h6]后乙烷转化率和o2残余含量(a)和乙烯生成选择性(b)的时间依赖性的绘图。
图9为显示在397℃在mo-v-te-nb-ox催化剂上气流切换[o2到c2h6]后乙烷转化率和o2残余含量(a)和乙烯生成选择性(b)的时间依赖性的绘图。
图10、11和12图示说明如何可用一系列三个固定床催化剂从氧化脱氢反应器中的产物流清除氧。
实施发明的最佳方式
数字范围:
除了操作实施例外,或者另外指明,说明书和权利要求书中使用的所有涉及成分的量、反应条件等的数字或表达均应理解为在所有情况下受词语“约”修饰。因此,除非指明为相反,以下说明书和附加权利要求中列出的数字参数均为近似值,可取决于本发明希望获得的所需性质变化。在最低限度,并且不作为尝试限制权利要求范围的等价原则应用,各个数字参数应至少按照所报告有效数字的数值并且通过运用普通舍入方法来解释。
尽管阐述本发明宽范围的数字范围和参数为近似值,但具体实施例中提到的数值均尽可能精确报告。尽管如此,由于在相应试验测量中发现的标准偏差,任何数值本身必然包含一定误差。
也应了解,本文陈述的任何最大数字范围旨在包括其中包含的所有子范围。例如,“1至10”的范围旨在包括在所陈述最小值1和所陈述最大值10之间,且包括二者,即具有等于或大于1的最小值和等于或小于10的最大值的所有子范围。由于所述数字范围是连续的,它们可包括最小值和最大值之间的每一个值。除非另外明确指明,在本申请中指定的不同数字范围为近似。
在本文中表达的所有组成范围在实际中总体限于且不超过100%(体积百分数或重量百分数)。在组合物中可存在多种组分时,各组分最大量的总和可超过100%,应理解,且本领域的技术人员容易理解,实际所用组分的量应符合最大100%。
催化剂:
有一些可根据本发明使用的催化剂。可单独或组合使用以下催化剂系统。本领域的技术人员应了解,应当以实验室规模试验这些组合,以确定是否在使用催化剂组合时有任何拮抗效应。
本发明的氧化脱氢催化剂可选自下列催化剂:
i)下式的催化剂:
nixaabbddoe
其中
x为0.1至0.9的数,优选0.3至0.9,最优选0.5至0.85,最优选0.6至0.8;
a为0.04至0.9的数;
b为0至0.5的数;
d为0至0.0.5的数;
e为满足催化剂价态的数;
a选自ti、ta、v、nb、hf、w、y、zn、zr、si和al或其混合物;
b选自la、ce、pr、nd、sm、sb、sn、bi、pb、tl、in、te、cr、mn、mo、fe、co、cu、ru、rh、pd、pt、ag、cd、os、ir、au、hg及其混合物;
d选自ca、k、mg、li、na、sr、ba、cs和rb及其混合物;且
o为氧;和
ii)下式的催化剂:
mofxgyh
其中
x选自ba、ca、cr、mn、nb、ta、ti、te、v、w及其混合物;
y选自bi、ce、co、cu、fe、k、mg、v、ni、p、pb、sb、si、sn、ti、u及其混合物;
f=1;
g为0至2;
h为0至2,其条件为对于co,ni,fe及其混合物的h的总值小于0.5;和
以下式iii)的催化剂,及
其混合物。
在一个实施方案中,催化剂为式i)的催化剂,其中x为0.5至0.85,a为0.15至0.5,b为0至0.1,且d为0至0.1。在催化剂i)中,一般a选自ti、ta、v、nb、hf、w、zr、si、al及其混合物,b选自la、ce、nd、sb、sn、bi、pb、cr、mn、mo、fe、co、cu、ru、rh、pd、pt、ag、cd、os、ir及其混合物,且d选自ca、k、mg、li、na、ba、cs、rb及其混合物。
在一个供选的实施方案中,催化剂为催化剂ii)。在本发明此方面的一些实施方案中,一般x选自ba、ca、cr、mn、nb、ti、te、v、w及其混合物,y选自bi、ce、co、cu、fe、k、mg、v、ni、p、pb、sb、sn、ti及其混合物。
一个另外的特别有用的催化剂系列iii)包括一种或多种选自下式混合氧化物催化剂的催化剂
vxmoynbztemmenop,
其中me为选自ti、ta、sb、hf、w、y、zn、zr、la、ce、pr、nd、sm、sn、bi、pb、cr、mn、fe、co、cu、ru、rh、pd、pt、ag、cd、os、ir、au及其混合物的金属;并且
x为0.1至3,优选0.5至2.0,最优选0.75至1.5,其条件为在me不存在时,x大于0.5;
y为0.5至1.5,优选0.75至1.0;
z为0.001至3,优选0.1至2,最优选0.5至1.5;
m为0.001至5,优选1至4;
n为0至2,优选n为0,然而,当me存在时,n优选为0.5至1.5;并且
p为满足混合氧化物催化剂价态的数。
在一个实施方案中,催化剂具有下式:
moavbnbcteeod
其中:
a为0.90至1.10,优选0.95至1.1;
b为0.25至0.4,优选0.3至0.35;
c为0.1至0.3,优选0.1至0.15;
e为0.1至0.3,优选0.1至0.25;并且
d为满足金属氧化物载体上混合氧化物催化剂价态的数。
在另一个实施方案中,在催化剂中,x:m之比为0.3至10,最优选0.5至8,理想0.5至6。
制备催化剂的方法为本领域的技术人员已知。
例如,可通过混合可溶金属化合物的水溶液制备催化剂,例如,氢氧化物、硫酸盐、硝酸盐、卤化物、低级(c1-5)单或二羧酸的盐和铵盐或金属酸本身。例如,可通过混合溶液制备催化剂,例如,偏钒酸盐铵、草酸铌、钼酸铵、碲酸等。然后一般在空气中在100-150℃干燥所得溶液,并在200-600℃(优选在300-500℃)在惰性气流中煅烧,例如,选自n2、he、ar、ne及其混合物的那些气流。煅烧步骤可进行1至20小时,一般5至15小时,通常约10小时。所得氧化物为一般不溶于水的易碎固体。
载体/粘合剂
有可负载或结合氧化脱氢催化剂的数种方式。
对于流化床和固定床反应器二者,用于形成陶瓷载体和用于粘合剂的优选组分包括钛、锆、铝、镁、硅的氧化物、磷酸盐、磷酸硼、磷酸锌及其混合物。在流化床中,一般催化剂利用粘合剂喷雾干燥,一般形成具有40-100um大小(有效直径)的球形颗粒。然而,需要仔细保证颗粒区域足够坚固,以使流化床中的磨损最大限度地减小。
用于固定床的催化剂的载体还可以为由选自二氧化硅、熔融二氧化硅、氧化铝、二氧化钛、二氧化锆、二氧化钍、氧化镧、氧化镁、氧化钙、氧化钡、氧化锡、二氧化铈、氧化锌、氧化硼、氮化硼、碳化硼、氧化钇、硅酸铝、氮化硅、碳化硅及其混合物的氧化物、二氧化物、氮化物、碳化物形成的陶瓷前体。
在一个实施方案中,对于氧化脱氢催化剂,用于固定床的载体可具有小于20m2/g的低表面积,或者小于15m2/g,或者小于3.0m2/g。这种载体可通过压缩模塑制备。在较高压力,陶瓷前体内的间隙被压塌。根据对载体前体施加的压力,载体的表面积可以为约20至10m2/g。
低表面积载体可以为任何常规形状,例如球、环、鞍等。
在本发明中,流化床中的经氧化催化剂包含一种或多种晶格氧和吸附氧。负载型催化剂与加入的空气或优选氧一起通过氧化脱氢反应器,且催化剂在乙烷转化成乙烯时被还原。然后,负载型催化剂由降管通到再生反应器,在此将其氧化。
重要的是在使用前(即,在加催化剂之前)使载体干燥。一般可在至少200℃温度加热载体最多24小时,一般在500℃至800℃温度加热约2至20小时,优选4至10小时。所得载体不含吸附水,且应具有约0.1至5mmol/g载体的表面羟基含量,优选0.5至3mmol/g。
二氧化硅上羟基的量可根据j.b.peri和a.l.hensley,jr.,在j.phys.chem.,72(8),2926,1968公开的方法测定,其全部内容通过引用结合到本文中。
然后,可通过压缩模塑将用于固定床催化剂的经干燥载体压缩成所需形状。根据载体的粒径,它可与惰性粘合剂混合,以保持压缩部分的形状。
负载量:
一般在用于固定床催化剂的载体上负载的催化剂分别提供1至30%重量所述催化剂(一般5至20%重量,优选8至15%重量)和99至70%重量所述载体(一般80至95%重量,优选85至92%重量)。
催化剂可以很多方式加到载体。例如,催化剂可通过浸渍、洗涂、刷或喷涂从含水浆料沉积于低表面积载体的表面之一上。催化剂也可与陶瓷前体(例如,氧化铝)从浆料共沉淀,以形成低表面积负载型催化剂。
用于流化床的催化剂负载量可基于多种因素选择,包括床体积、烷烃通过床的流速、床中的能量平衡、粘合剂类型等。对于流化床,催化剂负载量可覆盖10%重量至90%重量宽范围数值,一般高于20%重量,理想高于35%重量。
在本发明中,到氧化脱氢反应器的进料包括高于爆炸/燃烧上限量的氧。例如,对于乙烷氧化脱氢,氧一般以不小于约5%mol的量存在,优选约18%mol,例如约22至27%mol或23至26%mol。不希望有太高过量氧,因为这可由于进料或最终产物燃烧造成选择性减小。另外,在进料流中太高过量氧可能需要在反应下游端的另外的分离步骤。
结合图1描述本发明的方法,图1图示说明循环流化床反应器。
在一个实施方案中,反应器系统1包括流化床氧化脱氢反应器3和再生器反应器4。流化床氧化脱氢反应器升管2和再生反应器4由降管10连接,降管10将洁净的经氧化负载型催化剂从再生器反应器4传导到氧化脱氢反应器升管2。流化床氧化脱氢反应器升管2、脱氢反应器3中的流化床6、和再生反应器4分别包含催化剂颗粒流化床5、6和7。在氧化脱氢反应器升管2和再生反应器4中,脱离区域8和9分别高于流化催化剂床5和7。
降管10的入口11一般在流化床7高度约1/3至2/3之间的点结合到再生器反应器4。降管10进入氧化脱氢反应器升管2的底部。反应器升管2向上高于流化床6的流体平面伸入脱氢反应器3(一般为高度的1/3至2/3)。反应器升管2,张开形成倒锥形分散器12,以提供用于催化剂从产物的脱离区域。可任选高于锥12使用分散器板13。分散器可具有倒锥形以外的形状,然而,必须注意保证围绕分散器基本均匀的气流。
氧化脱氢反应器在低于450℃温度(一般350℃至450℃),在3.447至689.47kpa(0.5至100psi)压力(优选103.4至344.73kpa(15至50psi)),一种或多种烷烃在氧化脱氢反应器升管2中0.002至2秒的停留时间的条件下操作。
升管的张开区段12应足够宽,以使催化剂颗粒落入流化床区域6,且分散器板13应足够高,使得催化剂磨损最大限度地减小。
升管10中的口14允许在低于氧化脱氢反应器温度至少约25℃(期望50℃)的温度引入一个或多个低温蒸汽和雾化水(例如,雾)。在一些实施方案中,蒸汽具有约200℃至约400℃温度,在另一个实施方案中,温度可以为约300℃至350℃。蒸汽使来自再生器反应器4的催化剂冷却,也去除任何夹带或吸附的杂质(例如,乙烯或空气)。在引到口14时,雾化水可具有50℃至75℃温度。所有或部分雾化水可以为来自反应产物的再循环水。
在或朝向氧化脱氢反应器升管2底部的口15为烃进料的入口,烃进料一般为与氧或含氧气体混合的高纯度乙烷。烃进料和氧可接近氧化脱氢反应器和在其上游混合。由于这是流化床反应器,因此,必需使向上的烃进料和含氧气体流充分分布,流化催化剂颗粒床,使热点最大限度地减小。
可用本发明的方法从相对纯的原料产生乙烯。
乙烷应单独包含约95%重量乙烷(优选98%重量乙烷)和不大于约5%重量相关烃,例如甲烷。优选进料为具有相对高纯度的氧,在一些实施方案中高于90%纯度,在另一个实施方案中,高于95%纯度。虽然可用空气作为氧源,但它可造成下游分离问题。
在本发明的另一个实施方案中,可用本发明的反应器代替乙烷/乙烯分离器(splitter)或来自精炼厂或其它烃处理过程的排气,在此情况下,原料可包含10-80%体积乙烯和余量乙烷。
为了保持可用的流化床,通过床的质量气体流速必须高于流化所需的最低流速,优选约1.5至约10倍umf,更优选约2至约6倍umf。umf以接受形式作为达到流化所需最低质量气体流速的缩略使用,c.y.wen和y.h.yu,“mechanicsoffluidization”(流化机制),chemicalengineeringprogresssymposiumseries,vol.62,p.100-111(1966)。一般表观气体速度需要0.3至5m/s。
在氧化脱氢反应器上端,在脱离区域9下面有口16,口16允许已消耗催化剂流沉降并离开反应器。在反应器3顶部有旋风分离器17去除未在脱离区域9沉降的任何催化剂细屑。
负载型催化剂在氧化脱氢反应器升管2中的平均停留时间小于约30秒,在一些情况下小于15秒,在一些情况下1至6秒。口16连接降管18与氧化脱氢反应器3和再生反应器4。降管18中的口19布置接近再生反应器4。口19允许蒸汽在约300℃至500℃温度(在一些实施方案中350℃至450℃)相对于已消耗催化剂流逆流引入,以去除夹带的原料和产物。在一些情况下,蒸汽也可消除催化剂颗粒上的表面焦。降管中蒸汽的流速应低得足以防止负载型催化剂被推回到脱离区域9。
再生反应器也为流化床反应器。在再生反应器底部的口20允许空气和在一些情况下再循环的经冷却氮回到反应器中。再生反应器一般在250℃至400℃温度和3.447至689.47kpa(0.5至100psi)压力(优选103.4至344.73kpa(15至50psi))操作。负载型催化剂在再生反应器中的停留时间小于3分钟。一般地,催化剂在再生器反应器中的停留时间与在氧化脱氢反应器中的停留时间之比不小于3。
在再生器反应器4上部上高于负载型催化剂颗粒流化床的口21允许排气离开反应器。可如关于氧化脱氢反应器3中所述在再生器反应器4的上部区段有旋风分离器,以从氮产物流去除催化剂细屑。使可经冷却的空气和任选氮通过再生反应器。氧基本从空气提取出来。排气包含85至100%氮。
循环流化床反应器的以上描述大部分是示意性的。在口处可有各种阀、滤器等。选择适合阀为本领域的技术人员所熟悉。可类似具有用于强制气体通过系统的适合风机和压缩装置。选择适合风机或压缩机或膨胀机用于冷却为本领域的技术人员已知。
期望从氧化脱氢反应和再生反应回收尽可能多的能量。将乙烯进料和来自氧化反应器的副产物(例如,co2和co)送到单独的蒸汽发生器产生蒸汽。部分蒸汽可再循环回到过程中。可将蒸汽注入升管中,以冷却催化剂颗粒。也可将蒸汽注入降管,以消除任何焦炭,并夹带任何吸收或吸附的进料或产物。
通过再生器的含氧流在从反应器退出时基本耗减氧(例如,退出流包含不小于约90%氮)。如果还用氮作为进料流的组分,可使部分产物流再循环到再生反应器的入口。可使来自再生反应器的部分产物流经过一个或多个冷却或冷冻步骤,以保持再生器中热平衡。在一些实施方案中,可将co促进剂加到再生器,以使再生器中放热最大限度地减少(即,减少或控制co2产生)。
有一些专利和申请教导co促进剂,包括下面所列。
1977年12月20日颁予penick、转让给mobiloilcorporation公司的美国专利4,064,039教导加入最多50ppm(一般0.01至1ppm)铂族金属和铼,以促进co燃烧。
1978年2月7日颁予schwartz、转让给mobiloilcorporation公司的美国专利4,072,600教导加入1至50ppm选自铂、钯、铱、锇、钌和铼及其混合物的金属,以促进co燃烧。
1978年6月6日颁予schwartz、转让给mobiloilcorporation公司的美国专利4,093,535教导加入1至50ppm选自铂、钯、铱、锇、钌和铼及其混合物的金属,以促进co燃烧。
1996年10月15日颁予fraenkel等、转让给engelhardcorporation公司的美国专利5,565,399教导用于co促进剂的载体,所述载体包括已用至少2%重量la2o3和3至8%重量ceo2浸渍的氧化铝微球,所述微球基本不含α-氧化铝,并具有显示存在结晶ceo2的x-射线图案。
应操作本发明的方法,以具有不小于80%(到乙烯)的转化率,和不小于90%到乙烯的选择性,优选大于95%。
分离产物流:
退出脱氢反应的流22包含乙烯、水(蒸气-蒸汽)和少量乙烷、未消耗的氧和排气(一般co和co2)。在预期用于乙烯的背景下,需要考虑分离问题。
有一些方法可使用稀乙烯,例如聚合方法。然而,这种方法需要在极性分子(例如,co和co2及氧)对用于聚合的催化剂的影响方面取得平衡。可优选在分离乙烯和乙烷之前分离极性分子。极性分子可通过吸附床分离,例如沸石床。在最简单的实施方案中,根据组分比,可使床再生,并将所有组分送到燃烧器,以燃烧co。然而,在化学综合体,有其它单元操作可用co作为进料(各种羧酸和酐过程(乙酸、甲基丙烯酸和马来酸酐)。如果有显著量co和co2,则可使组分分离。在本领域有一些分离co2和co的熟知方法。冷却并洗涤该流,然后使其通过吸附器,例如活性炭(以从co2去除杂质),或通过液体胺分离器或液体碳酸盐分离器,以吸附co2。可通过一些技术分离co。根据体积,用活性炭作为吸附剂的真空分离方法可能适合,膜分离可能适合,在铜离子上吸附(在适合载体上)可能适合。
除氧-固定床:
在一个实施方案中,可有两个或更多个具有释放或吸收氧的氧化脱氢催化剂的固定床反应器,用作清除器,以容纳从循环流化床氧化脱氢反应器出来的产物流。固定床反应器具有管和阀,以使到流化床氧化脱氢反应器的进料通过一个或多个固定床,所述固定床具有包含消耗或放出的氧的催化剂。这对于在氧化脱氢模式操作的预反应器不是太大问题,因为在预反应器中未脱氢的任何过量烷烃将在流化床氧化脱氢反应器中转化。关键问题是使固定床反应器中的催化剂耗减氧。管和阀使产物流通过具有耗减氧的氧化脱氢催化剂的一个或多个固定床反应器。耗减的固定床催化剂从产物流清除氧。如前提到,可操作物流的阀和管,以使进料流流动通过含氧固定床催化剂反应器,且产物流流动通过一个或多个耗减氧的固定床催化剂反应器。
含氧的氧化脱氢催化剂可在催化剂、载体或二者上具有氧作为晶格氧、吸附氧或吸收氧。耗减氧的氧化脱氢催化剂具有在催化剂和载体中减少(优选少约60%)的氧作为晶格氧、吸附氧或在催化剂、载体或二者上的吸附氧。
优选在流化床氧化脱氢反应器的出口有氧传感器。另外,在来自各固定床反应器的脱氢产物的出口应有氧传感器,以测定离开该固定床反应器的产物的氧含量。在以清除器模式操作的固定床反应器的脱氢产物出口氧含量升高时,表明催化剂已基本吸收反应性氧(并且可返回用作预反应器)。在氧清除或化学吸附模式的预反应器操作中由氧耗减催化剂吸收的反应性氧的量应不小于催化剂中总氧的约1.5%,一般约2%(这也对应于可从氧化脱氢模式预反应器中催化剂释放的反应性氧的量)。
用三个预反应器操作的一种模式图示说明于图10、11和12(其中相似部件具有相似数字)和下表中。在图10、11和12中未显示阀。主反应器结构相同,然而,阀的切换使预反应器、清除器反应器和保护反应器似乎“切换”位置。一个预反应器如此操作,并使部分进料流转化成乙烯。一个氧耗减预反应器作为主氧清除器或化学吸附反应器和第二预反应器(氧耗减反应器也作为保护或第二氧清除器或化学吸附反应器)。
操作:
在供选实施方案中,可用低温方法从产物流分离氧。然而,这增加过程的资本和操作成本。
以上清除方法更充分描述于2013年11月21日提交的加拿大专利申请2,833,822,其文本通过引用结合到本文中。
也使来自降管的残余气体经过相同分离技术,以回收它们。
如上提到,可不必在此阶段从乙烯分离乙烷,然而,如果需要,有一些可用的技术。
最普通的技术是使用低温c2分离器。其它分离技术包括下列。
分离产物流的一种方法是通过吸收。主要包含乙烷和乙烯的气态产物流可以逆流与较重石蜡油(例如,矿质海豹油或药用白油)在最高800psi(约5.5x103kpa)压力和约25℉至125℉(约-4℃至约52℃)温度接触。乙烯和较低沸点组分不被吸收到油中。乙烷和较高沸点组分吸收到油中。如果需要,乙烯和较低沸点组分然后可通到c2分离器。吸收油可选择性用溶剂萃取,例如,糠醛、二甲基甲酰胺、二氧化硫、苯胺、硝基苯和用于萃取任何较重链烷烃的其它已知溶剂。这种方法更充分描述于1945年5月15日颁予welling、转让给phillipspetroleumcompany公司的美国专利2,395,362,其内容通过引用结合到本文中。
另一种分离方法为吸附方法。吸附剂优先吸附产物流中的组分之一。吸附方法一般包括两个或更多个吸附单元的列,以便在单元已达到负载能力时,将进料引到替代单元,同时再生完全负载单元,一般通过以下一个或多个因素:温度或压力或二者的变化。
有相当多技术用+1氧化态的银或铜离子分离乙烯和乙烷。烯烃优先吸附进入包含选自溶于溶剂的银(i)或铜(i)盐的络合剂的络合溶液。一些银吸附剂包括硝酸银、氟硼酸银、氟硅酸银、羟基氟硼酸银和三氟乙酸银。一些铜吸附剂包括硝酸亚铜;卤化亚铜,例如氯化亚铜;硫酸亚铜;磺酸亚铜;羧酸亚铜;氟代羧酸的亚铜盐,例如,三氟乙酸亚铜和五氟乙酸亚铜;氟化乙酰丙酮根合亚铜;六氟乙酰丙酮根合亚铜;十二烷基苯磺酸亚铜;卤化铜铝,例如四氯化亚铜铝;cualch3cl3;cualc2h5cl3;和氰基三氯化亚铜铝。如果在与液体吸附剂接触之前已干燥产物流,吸附剂应对水解稳定。络合剂优选稳定,并在溶剂中具有高溶解性。在一个吸附剂溶液被基本负载后,将产物流进料切换到另一溶液。然后,通过热或压力或二者的变化再生完全负载的吸附剂溶液。这释放乙烯。
这些类型的方法描述于2003年2月11日颁予culp等、转让给unioncarbidechemicals&plasticscorporation公司的美国专利6,581,476和1999年1月12日颁予barchas等、转让给stoneandwebsterengineering的美国专利5,859,304,其内容通过引用结合到本文中。
在溶液方法的替代方法中,载体,例如沸石4a、沸石x、沸石y、氧化铝和二氧化硅,可用铜盐处理,以从包含饱和烃(即,链烷烃,如乙烷和丙烷)的气体混合物选择性去除一氧化碳和/或烯烃。1990年4月17日颁予xie等、转让给北京大学的美国专利4,917,711描述使用这种负载型吸附剂,其内容通过引用结合到本文中。
类似地,2005年3月15日颁发的美国专利6,867,166和2002年7月23日颁予yang等、转让给regentsoftheuniversityofmichigan的美国专利6,423,881,通过引用结合到本文中,描述用供选负载于二氧化硅、氧化铝、mcm-41沸石、4a沸石、碳分子筛、聚合物(例如amberlyst-35树脂)和氧化铝上的铜盐和银化合物,选择性从包含烯烃和链烷烃的气体混合物吸附烯烃。观察和模型化动力学和热力学分离两种性质。吸附烯烃在1至35个大气压压力(优选小于10个大气压,最优选小于2个大气压)在0至50℃温度(优选25至50℃)进行,解吸在0.01至5个大气压压力(优选0.1至0.5个大气压)在70℃至200℃温度(优选100℃至120℃)进行。
在另一个实施方案中,吸附剂可以为选自天然和合成沸石的物理吸附剂,没有银或铜盐。
一般地,吸附剂可以为氧化铝、二氧化硅、沸石、碳分子筛等。典型吸附剂包括氧化铝、硅胶、碳分子筛、沸石(如a型和x型沸石、y型沸石)等。优选的吸附剂为a型沸石,最优选的吸附剂为4a型沸石。
4a型沸石,即,钠型a型沸石,具有约3.6至4埃单位的表观孔径。这种吸附剂提供在升高温度从乙烯-乙烷混合物吸附乙烯和从丙烯-丙烷混合物吸附丙烯的提高选择性和容量。在基本未改性时,即,在只有钠离子作为其可交换阳离子时,该吸附剂最有效用于本发明。然而,吸附剂的某些性质,例如热和光稳定性,可通过一些钠离子与其它阳离子(除银或铜之外)部分交换来改善。因此,使用4a型沸石在本发明的优选实施方案范围内,其中结合到吸附剂的一些钠离子用其它金属离子置换,其条件为交换的离子的百分数不大到吸附剂损失其4a型特性。在限定4a型特性的性质中,有吸附剂在升高温度选择性从乙烯-乙烷混合物吸附乙烯和从丙烯-丙烷气体混合物吸附丙烯,且达到此结果而不导致混合物中存在的烯烃显著低聚或聚合的能力。一般已确定,4a沸石中最高约25%(按当量计)钠离子可通过离子交换用其它阳离子置换,而不夺去吸附剂的4a型特性。可与烯烃-烷烃分离中所用4a沸石离子交换的阳离子尤其包括钾、钙、镁、锶、锌、钴、锰、镉、铝、铈等。当以其它阳离子交换钠离子时,优选小于约10%钠离子(按当量计)被这些其它阳离子置换。置换钠离子可改变吸附剂的性质。例如,用其它阳离子取代一些钠离子可改善吸附剂的稳定性。如1998年4月28日颁予ramachandran等、转让给bocgroup,inc.公司的美国专利5,744,687所公开,其内容通过引用结合到本文中。
特别优选的沸石为zsm-5。
除了沸石外,还有一些钛硅酸盐同系物,也称为ets化合物。
美国专利5,011,591公开合成大孔径钛硅酸盐,也称为“ets-10”。与ets-4和cts-1(以下提及)相比,大孔钛硅酸盐材料ets-10,具有约8å孔径,不能在动力学上区分轻烯烃与相同碳数的链烷烃。然而,已报告用制备态ets-10沸石从乙烷分离乙烯的高度选择性;见al-baghli和loughlin,j.chem.eng.data2006,v51,p248。作者证明,na-ets-10能够在热力学条件下甚至在环境温度从乙烯和乙烷的混合物选择性吸附乙烯。虽然关于用na-ets-10吸附乙烯的报告选择性在环境温度高,但乙烯和乙烷的吸附等温线具有与低变压容量一致的高度呈长方形的形状。因此,na-ets-10不容易应用于变压吸附过程(psa),至少在较低或环境温度。
然而,阳离子改性制备态na-ets-10提供在环境温度psa分离具有相同碳原子数烯烃和链烷烃的吸附剂。单、二和三价阳离子选自第2-4族金属、质子、铵化合物及其混合物。可用于本发明的单、二或三价阳离子的一些具体非限制实例包括li+、k+、cs+、mg2+、ca2+、sr2+、ba2+、sc3+、y3+、la3+、cu+、zn2+、cd2+、ag+、au+、h+、nh4+和nr4+,其中r为烷基、芳基、烷基芳基或芳基烷基。离子改性剂一般以盐或酸的形式加到未改性na-ets-10。与阳离子改性剂相关的阴离子反离子未明确限定,其条件为不有害影响改性(即,阳离子交换)反应。适合阴离子包括但不限于乙酸根、羧酸根、苯甲酸根、溴酸根、氯酸根、高氯酸根、亚氯酸根(chorite)、柠檬酸根、硝酸根、亚硝酸根、硫酸根和卤离子(f,cl,br,i)及其混合物。适合的酸包括无机和有机酸,无机酸是优选的。2011年9月13日颁予kuznicki等、转让给governorsoftheuniversityofalberta的美国专利8,017,825公开该技术,其文本通过引用结合到本文中。
如美国专利6,517,611中所述,热处理ets-4得到受控孔体积沸石材料,称为“cts-1”,这是用于烯烃/链烷烃分离的高选择性吸附剂。cts-1沸石,具有约3-4å孔径,通过尺寸排阻过程从乙烯和乙烷的混合物选择性吸附乙烯。cts-1的孔径允许乙烯扩散,同时阻止太大而不能进入cts-1沸石孔的乙烷扩散,从而提供动力学分离。cts-1吸附剂成功应用于psa过程,其中可分别从乙烷或丙烷分离乙烯或丙烯。
以上吸附剂可用于变压吸附单元。一般在吸附步骤期间使用的绝对压力范围可以为约10kpa至约2,000kpa(约1.5至约290磅/平方英寸(psi)),优选约50kpa至约1000kpa(约7.2至约145psi)。在释放被吸附物期间(即,在再生步骤期间)使用的压力范围可以为约0.01kpa至约150kpa(约0.0015至约22psi),优选约0.1kpa至约50kpa(约0.015至约7.3psi)。一般地,吸附步骤可在环境温度至高于约200℃进行,优选低于150℃,最优选低于100℃,其条件为温度不超过烯烃化学反应(例如,低聚或聚合)进行的温度。
另一类吸附剂为离子液体。烯烃和链烷烃可用选自下式的络合物的金属二硫纶的离子液体分离:
(i)m[s2c2(r1r2)]2;
和
(ii)m[s2c6(r3r4r6r7)]2.
其中m选自fe、co、ni、cu、pd和pt;r1、r2、r3、r4、r5和r6独立选自氢原子、吸电子基团(包括为或包含杂环、氰基、羧酸盐、羧酸酯、酮基、硝基和磺酰基的那些基团)和选自c1-6烷基、c5-8烷基、c2-8烯基和c6-8芳基的烃基,其中烃基未取代或完全或部分取代,优选由卤素原子取代的那些烃基。离子液体可与非反应性溶剂或助溶剂一起使用。溶剂可选自常规芳族溶剂,一般为甲苯。吸附压力可以为200psig至300psig(1.3x103至2x103kpag),优选低于250psig(1.7x103kpag),吸附温度可以为环境温度至200℃,优选低于150℃,烯烃可从离子液体通过以下一种或多种方式来释放:降低压力至少50psig(3.4x102kpa)和升温不小于15℃。
可在一些下游单元操作中使用来自再生反应器而未循环到再生反应器的氮。可能的下游单元操作包括氨装置、丙烯腈装置、脲装置和硝酸铵装置。
以下非限制实施例说明本发明。可在试验中使用的催化剂具有下式:
moavbnbcteeod
其中:
a为0.90至1.10;
b为0.25至0.4;
c为0.1至0.3;
e为0.1至0.3;并且
d为满足混合氧化物催化剂价态的数。
在试验中使用的反应器由石英管反应器组成。样品尺寸一般为约0.5cm3,0.17g。催化剂的粒径为0.2-0.7mm。
反应器最初以再生(催化剂氧化)模式操作。将反应器在空气中加热到355℃至397℃经历30分钟。然后将气流切换到75%体积乙烷和25%体积氧的混合物。乙烷和氧的混合物的流速在300/600/1200cm3(stp)/小时内变化。反应在反应剂通过经氧化催化剂床的第一分钟期间进行。然后再氧化催化剂床,然后使乙烷和氧的混合物通过经氧化催化剂。分析离开反应器的气体,以检测残余氧和产物气体中乙烷、乙烯和副产物的量。
试验#1:
(空气
图2和3显示通过在600cc/hr供应的反应混合物在两个不同温度预氧化催化剂逐渐还原时乙烷和o2转化率以及乙烯生成选择性的时间依赖性。
可以看到(图2,3),所有瞬时过程在我们的试验条件中在第一分钟操作期间发生。在~355℃效果不明显,可注意到在没有任何选择性损失下转化率只略微增加(图2)。在400℃,相同转化率升高效果变得强得多,但在此情况下,由于另外生成co2,伴随选择性显著损失(图3)。必须注意到,在400℃更活跃发生的过程伴随催化剂层的可检测自热(在反应器壁上测量~5-6℃)。不能排除在气相中不合需要的完全氧化的一些贡献。
试验#2:
在398℃重复试验1。
比较试验1和2,最大转化率增加到高于70%。
试验#3:
为了阐明本发明,使用与实施例1和2相同的条件,利用不同气体流速(300和1200cc/min)进行另外的试验,不同之处在于流速为1200cm3/h。所得结果提供于图4和5中。
所得数据(图3-5)显示,选择性与进料流速(空间速度)相关。气体流速减到600h-1使选择性临时下降到75%(图5b)。同样,该过程在气体切换到反应混合物后伴随显著催化层显著自热(在反应器壁上测量~6-7℃)。选择性曲线概括并比较于图6中。引人关注的是,注意到在短停留时间,尽管有高转化率,但消耗很少气相氧。因此,在增加接触时间时,在临时加热催化剂床的情况下,不合需要的完全氧化的贡献变得越来越明显(图6)。同时,气体流速增加到2400h-1使我们避免显著总氧化贡献(图6)。
为了定量比较转化率数据,用绝对标度在图7中提供在相差2倍流速下得到的所有结果(即,作为送过反应器的乙烷的量的函数)。所有三条曲线看起来很相似(图7)。显然,由在预氧化催化剂中储存的额外氧存在导致增加起始乙烷转化率(70-80%),图7中所示的瞬时过程与这种额外氧的逐渐损失相关。所得结果使我们能够计算在瞬时过程期间反应中包括的“反应性”晶格氧的量。对于所有三个试验,从催化剂耗减氧是相同的,并且可从我们的mo-v-te-nb-ox催化剂的总晶格氧评估为~1%。
试验#4:
周期氧化还原循环(纯o2
为了阐明本发明,在相同的其它条件下(即,流速和温度),在乙烷流中在无氧存在下进行本试验。在此试验中,将放入石英反应器的催化剂装料在纯o2流中加热到指定温度(354℃或397℃),保持30min,使气流(600cm3/h)切换到纯c2h6,并在指定时间后分析引出气体样品。催化剂再氧化30min后,用不同时间间隔重复检测数次,接收到产物所得的响应曲线(多达3min)。图8和9显示在两个不同温度通过乙烷使催化剂逐渐还原时,乙烷转化率和残余o2含量及乙烯生成选择性的时间依赖性。
瞬时过程在我们的试验条件(图8,9)在1-2分钟期间发生。反应伴随可检测选择性损失。效果甚至在~350℃也很明显(图8b),在400℃变得更强(图9b)。重要的是,应注意到,反应伴随催化剂层的可检测自热(在反应器外壁上测量4-8℃)。切换回到o2流用于催化剂再氧化也伴随一些催化剂加热(3-4℃)。另外,此加热似乎不均匀,但在反应期间在整个层移动。考虑到这种催化剂过度加热在催化剂床内显著更强,由在两种纯气体之间切换提供的非等温条件作用可能是重要的。
以上实施例也说明,使用odh脉冲模式的转化率和选择性不如本发明有效。
工业应用
本发明提供使用循环床反应器(类似于流化床反应器裂化器(fcc))的氧化脱氢方法,该方法以高选择性提供良好的烯烃产物产率。
- 还没有人留言评论。精彩留言会获得点赞!