DNA展示及其方法与流程
1.引言
本公开总体上涉及dna展示、制备dna展示文库的方法、使用dna展示或dna展示文库以选择与靶标结合的配体的方法。本文描述了制备用于展示从dna模板转录的rna的dna展示文库的方法。该方法包括通过桥连接头分子将三磷酸核苷(rna)连接到寡核苷酸(dna)的过程。本文还描述了捕获转录产物(rna)并将其连接到其编码模板(dna)的方法。本文还描述了通过其编码模板(dna)将转录产物(rna)与支架例如固体支持物或珠连接的方法。本文描述了将通过其编码转录产物(rna)的翻译产物(例如但不限于蛋白质、肽、抗体或酶)通过其编码模板(dna)连接到支架例如固体支持物或珠的方法。在具体实施方案中,选择是结合配体例如rna适体和蛋白质的体外选择。本文还描述了包含dna展示的试剂盒。本文还公开了包含与dna展示编码链互补的dna展示模板链的双链dna-rna融合物,只是所述dna展示编码链在其5'末端还包含接头分子和rna分子,并且其中所述rna分子从dna展示编码链转录。本文公开了包含双链dna-rna融合物的组合物,以及其制备方法及其在选择靶标结合配体中的用途。
2.
背景技术:
有几个展示平台旨在实现基于其功能展示和分离dna和蛋白质的目标。在过去20年中,使用selex(us5696249a)的dna和rna适体的体外选择已经产生高亲和力配体。这种类型的体外选择与使用的体内选择方法相比具有一些明显的优点,主要是能够被筛选的大的文库大小(>1015个分子)(osborne和ellington1997)。
体内选择技术例如酵母展示(us6699658b1)的一个优点是使用荧光激活细胞分选(facs)进行选择的能力。这显著增强了选择属性的范围和选择的准确性(vanantwerp和wittrup2000)。酵母展示是非常成功的体外抗体选择策略,主要是由于facs分选。酵母展示和其它细胞表面展示技术(us5348867a)的缺点是候选文库大小受限于所用生物体的转染效率,酵母展示的文库大小限制为约1010个分子(benatuil,perez等人,2010)。
最近开发了粒子展示技术(wang等,2014),其利用乳液pcr在珠上展示整个dna适体文库,每个珠展示一个适体序列的多个拷贝。这允许dna适体的荧光激活细胞分选(facs),产生比先前的dna适体选择方法强1000倍的结合亲和力(wang等人,2014)。
目前在本领域中可用和使用的分子展示技术是mrna展示(liu,roberts等人2000)(us6261804b1)、核糖体展示(he和taussig1998)(us6620587b1)、噬菌体展示(scott和smith1990)(sidhu,weiss等人2012)(us8685893b2)、酵母展示(wittrup,kranz等人2004)(us6699658b1)、细菌展示(earhart,francisco等人1993)(us5348867a)和cis展示(coomber,eldridge等人2004)(us8679781b2)。
mrna展示(us6261804b1),其是蛋白质展示技术,利用嘌呤霉素(酪氨酰-trna和腺苷的模拟物),通过接头连接到mrna序列的末端,并且在翻译过程中将嘌呤霉素插入到形成的蛋白质,产生mrna与mrna编码的蛋白质之间的共价连接。
核糖体展示(us6620587b1),其是蛋白质展示技术,涉及在将mrna翻译成蛋白质的结束时停止核糖体。这是由于去除了mrna终止密码子和加入了限制核糖体沿着mrna运动的高镁浓度的缓冲液。
噬菌体展示(us8685893b2),其是蛋白质展示技术,使用经遗传工程化的噬菌体,使得其外壳蛋白基因包括文库序列。这意味着噬菌体在外部展示蛋白质,同时在内部含有dna和rna遗传信息。
酵母展示(us6699658b1),其是蛋白质展示技术,涉及在酵母细胞表面上将目的蛋白质展示为具有aga2p的融合蛋白。遗传信息rna和dna都包含在酵母细胞内。
细菌展示(us5348867a),其是蛋白质展示技术,涉及将目的蛋白质表达为具有天然表面展示蛋白的融合蛋白。
cis展示(us8679781b2),其是蛋白质展示技术,利用蛋白repa,一种与表达该蛋白的dna模板结合的dna复制起始蛋白(mcgregor等人,2003)。使用repa,cis展示将蛋白质与蛋白质所来源的dna结合。cis展示有一些缺点。所需的repa蛋白长度为351个氨基酸残基(dna序列为1053bp),需要在具有文库的融合蛋白内。这是一个很大的尺寸,所以增加立体位阻。此外,cis展示中使用的dna与蛋白质之间的连接是非共价的,因此易于分离。使用cis展示的任何基于捕获的选择将具有结合亲和力的上限,所述结合亲和力是repa/dna相互作用强度。这是cis展示的主要缺点。
粒子展示,其是dna展示技术,涉及将引物与磁珠缀合。然后可以使用乳液pcr将文库dna扩增到珠上,该方式使得每个珠展示单个dna适体序列的约105个拷贝(wang,gong等人,2014)。
以前显示的展示技术通常展示蛋白质。不存在展示rna的技术。需要一种允许rna展示在dna上的新平台。
3.
技术实现要素:
本文描述了一种在dna上展示rna以及应用于rna的体外选择和体外评价的快速有效的方法。本公开有利于从部分或完全随机dna序列的大库中分离具有所需性质的rna。此外,本发明通过将rna分子共价连接到其编码dna来消除转录完成后的空间信息丢失。通过dna展示模板进行dna展示可用于在转录后捕获rna。这种捕获的应用可以是基于研究的,如转录产物的定量,或更多应用,如将基因型(dna)与表型(rna)相关联用于rna适体和蛋白质的体外进化。重要的是,dna展示连接另外两种技术,即粒子展示和mrna展示。跨接这个间隙意味着蛋白质,例如抗体或酶,可以在珠粒上选择,而在没有dna展示的情况下这是不可能的。这在蛋白质定向进化中具有重要应用,其产生用于清洁产品、食品工业、生物催化剂替代能量生产、医疗用途、生物化学和许多其它工业的抗体和酶。
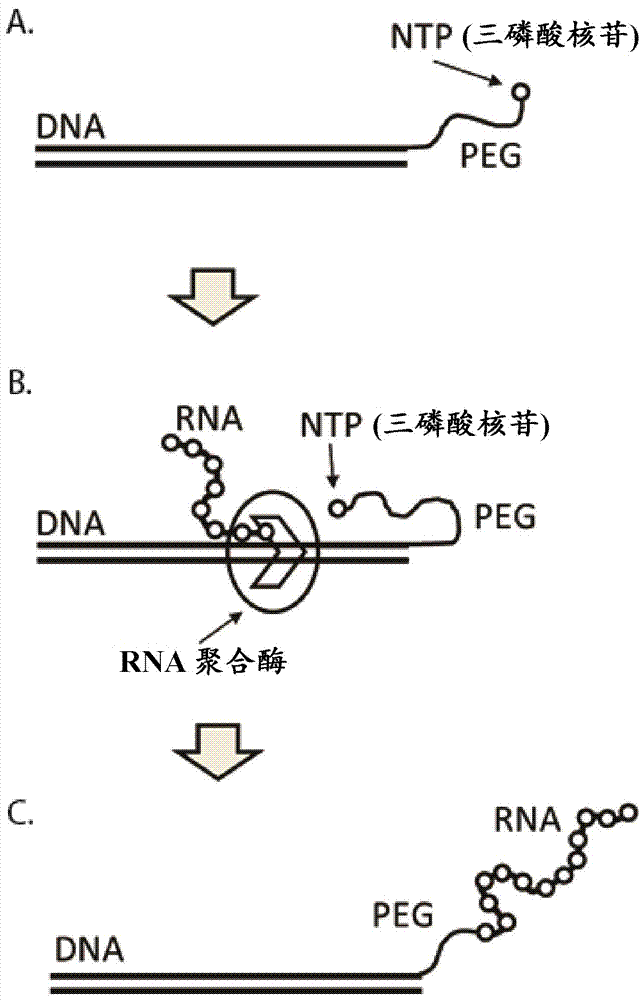
dna展示是用于将新转录的rna共价连接到其相应的dna模板的新技术。这是通过使用dna展示模板来实现的。dna展示模板通过将三磷酸核苷(ntp)与dna模板通过如图1所示的柔性peg接头缀合来制备。通常,该方法由体外或原位延伸/转录方案组成,这产生共价连接到编码rna的dna的3'末端的rna,即dna-rna融合物。在一个实施方案中,dna展示模板接头分子(dna-接头-ntp)利用peg作为接头形成dna展示模板peg分子(dna-peg-ntp)。在该实施方案中,peg通过peg上的n-羟基琥珀酰亚胺(nhs)官能团与ntp上的氨基烯丙基反应而与ntp缀合。然后通过peg上的马来酰亚胺官能团与模板dna上的硫醇官能团反应将ntp-peg缀合物与模板dna缀合。最终产物是如图1所示的dna-peg-ntp缀合物。然后可以将该dna-peg-ntp缀合物用于在转录过程中捕获rna。
当dna展示模板被转录时,通过rna聚合酶将缀合ntp插入到新形成的rna中,产生模板dna与其编码的rna之间的共价连接,如图3所示,这形成dna-rna融合物。
该rna捕获过程可以在选择轮次中重复。来自所选择的dna-rna融合物的dna可以被扩增、ntp修饰和转录以产生准备用于下一轮选择的初始dna-rna融合物。进行多轮选择和扩增的能力使得能够富集和分离非常罕见的分子,例如1015个成员库中的一个所需分子。这继而允许分离新的或改进的rna或rna适体,其特异性地识别几乎任何靶标或催化所需的化学反应。
因此,一方面,本发明的特征在于选择所需rna的方法,其包括以下步骤:(a)提供候选dna分子群体,其各自包含正向引物位点、与候选rna编码序列可操作地连接的t7rna聚合酶启动子,其中各自可操作地连接到具有接头-ntp修饰的反向引物位点;(b)体外或原位转录候选rna编码序列以产生候选dna-rna融合物群体;和(c)选择所需的dna-rna融合物,从而选择所需的rna。
在相关方面,本文描述了选择编码所需蛋白质的dna分子的方法,其包括以下步骤:(a)提供候选dna分子群体,其各自包含正向引物位点、与候选rna编码序列可操作地连接的t7rna聚合酶启动子,并且其各自可操作地连接到具有接头-ntp修饰的反向引物位点;(b)体外或原位转录候选rna编码序列以产生dna-rna融合物群体;和(c)用嘌呤霉素连键修饰rna的5'末端;和(d)体外或原位翻译候选rna以产生候选dna-rna-蛋白质融合物群体;和(e)选择所需的蛋白质,从而选择所需的dna-rna-蛋白质融合物。
在一个方面,本文描述了制备展示rna的dna展示文库的方法。
一方面,本文描述了生产dna展示文库的方法,包括:(i)提供dna编码链的群体,其各自包含正向引物位点、t7rna聚合酶启动子、dna编码区和反向引物结合位点;(ii)将每个dna编码链退火至正向引物;(iii)在dna聚合酶存在下延伸引物以形成双链dna展示模板群体;(iv)使双链dna展示模板群体变性以形成单链dna展示模板链群体;(v)将包含反向引物的ntp-接头-dna缀合物退火至每个单链dna展示模板链;和(vi)在dna存在下通过pcr延伸ntp-接头-dna缀合物的反向引物,以形成包含接头-ntp的双链dna展示模板群体。
在某些实施方案中,ntp-接头-dna缀合物通过包括以下步骤的方法产生:(i)将氨基烯丙基三磷酸核苷(aa-ntp)与n-羟基琥珀酰亚胺(nhs)-peg-马来酰亚胺(nhs-peg)缀合,形成ntp-peg-马来酰亚胺(ntp-peg);(ii)将用还原硫醇官能化的dna寡核苷酸(硫醇-dna)与ntp-peg上的马来酰亚胺缀合,形成ntp-peg-dna;和(iii)纯化ntp-peg-dna缀合物。
在某些实施方案中,ntp-接头-dna缀合物包含长度为200-300埃的接头。在某些实施方案中,接头不包含多核苷酸。
一方面,本文提供了生产dna展示模板包被珠的方法,包括:(i)提供包含正向引物位点、t7rna聚合酶启动子、编码区和反向引物结合位点的dna展示文库编码链;(ii)提供正向引物缀合珠;(iii)将dna展示文库编码链退火至正向引物缀合珠;(iv)在dna聚合酶存在下,在正向引物缀合珠上将引物延伸,形成包含双链dna展示模板的珠;(v)使双链dna展示模板变性以形成包含单链dna展示模板链的珠;(vi)将包含反向引物的ntp-peg-dna缀合物与单链dna展示模板链退火;(vii)在dna存在下通过pcr延伸ntp-peg-dna缀合物的反向引物以形成包含具有peg-ntp的双链dna展示模板的珠。
在一个实施方案中,通过以下步骤产生ntp-peg-dna缀合物:(i)将氨基烯丙基三磷酸核苷(aa-ntp)与n-羟基琥珀酰亚胺(nhs)-peg-马来酰亚胺(nhs-peg)缀合,形成ntp-peg-马来酰亚胺(ntp-peg);(ii)将用还原硫醇官能化的dna寡核苷酸(硫醇-dna)与ntp-peg上的马来酰亚胺缀合,形成ntp-peg-dna;和(iii)纯化ntp-peg-dna缀合物。
在一个实施方案中,正向引物缀合珠由羧酸官能化珠与胺官能化正向引物之间形成的氨基键形成。
一方面,本文提供了生产ntp-peg-dna缀合物的方法,包括:(i)将氨基烯丙基三磷酸核苷(aa-ntp)与n-羟基琥珀酰亚胺(nhs)-peg-马来酰亚胺(nhs-peg)缀合,形成ntp-peg-马来酰亚胺(ntp-peg);(ii)将用还原硫醇官能化的dna寡核苷酸(硫醇-dna)与ntp-peg上的马来酰亚胺缀合,形成ntp-peg-dna;和(iii)纯化ntp-peg-dna缀合物。
在一个实施方案中,aa-ntp是氨基烯丙基三磷酸胞苷。
在一个实施方案中,nhs-peg是n-羟基琥珀酰亚胺马来酰亚胺。
在一个实施方案中,还原硫醇是还原的5’c6s-s硫醇修饰的dna寡核苷酸。
在一个实施方案中,ntp-peg-dna缀合物是ctp-peg-dna缀合物。
在一个实施方案中,pcr是乳液pcr,其中每个珠展示单个编码链序列的多个拷贝。
在一个实施方案中,乳液被破坏,并且珠上的双链dna变性为单链dna。
在一个实施方案中,dna编码链是反向引物。
一种制备包含rna适体的文库的方法,所述方法包括以下步骤:(i)提供dna编码链的群体,其各自包含正向引物位点、t7rna聚合酶启动子、编码区和反向引物结合位点;(ii)将每个dna编码链退火至正向引物;(iii)在dna聚合酶存在下延伸正向引物以形成双链dna展示模板群体;(iv)使双链dna展示模板群体变性以形成单链dna展示模板链群体;(v)将包含反向引物的ntp-peg-dna缀合物退火至每个单链dna展示模板链;和(vi)通过pcr在dna存在下延伸ntp-peg-dna缀合物的反向引物以形成具有peg-ntp的双链dna展示模板群体。
在一个实施方案中,所述ntp-peg-dna缀合物通过以下步骤产生:(i)将氨基烯丙基三磷酸核苷(aa-ntp)与n-羟基琥珀酰亚胺(nhs)-peg-马来酰亚胺(nhs-peg)缀合,形成ntp-peg-马来酰亚胺(ntp-peg);(ii)将用还原硫醇官能化的dna寡核苷酸(硫醇-dna)与ntp-peg上的马来酰亚胺缀合,形成ntp-peg-dna;(iii)纯化ntp-peg-dna缀合物;和(iv)在包含双链dna展示模板的珠上将dna展示模板体外转录。
在一个方面,本文提供了制备包含rna适体的文库的方法,所述方法包括以下步骤:(i)提供dna编码链的群体,其各自包含正向引物位点、t7rna聚合酶启动子、编码区和反向引物结合位点;(ii)提供正向引物缀合珠;(iii)将每个dna展示编码链退火至正向引物缀合珠;(iv)在dna聚合酶存在下将正向引物缀合珠上的引物延伸以形成双链dna展示模板群体;(v)使双链dna展示模板群体变性以形成单链dna展示模板链群体;(vi)将包含反向引物的ntp-peg-dna缀合物与每个单链dna展示模板链退火;(vii)在dna聚合酶存在下通过pcr延伸ntp-peg-dna缀合物的反向引物以形成具有peg-ntp的双链dna展示模板群体。
在一个实施方案中,所述ntp-peg-dna缀合物通过以下步骤产生:(i)将氨基烯丙基三磷酸核苷(aa-ntp)与n-羟基琥珀酰亚胺(nhs)-peg-马来酰亚胺(nhs-peg)缀合,形成ntp-peg-马来酰亚胺(ntp-peg);(ii)将用还原硫醇官能化的dna寡核苷酸(硫醇-dna)与ntp-peg上的马来酰亚胺缀合,形成ntp-peg-dna;(iii)纯化ntp-peg-dna缀合物;和(iv)在包含双链dna展示模板的珠上将dna展示模板体外转录。
在一个实施方案中,正向引物缀合珠由羧酸官能化珠与胺官能化正向引物之间形成的氨基键形成。
在一个方面,本文提供了筛选rna适体的方法,包括:(i)将包含rna适体的珠与标记的靶蛋白一起孵育足以使rna适体与靶蛋白结合的时间;(ii)洗涤以除去未结合的rna适体;(iii)选择与靶蛋白结合的rna适体。
在一个方面,所述方法还包括以下步骤:(i)使用pcr扩增所选择的rna适体的dna模板;和(ii)重复数轮pcr扩增,其足以对文库测序以鉴定分离的适体。
在一个方面,所述方法还包括以下步骤:(i)使用pcr扩增所选择的rna适体的dna模板;(ii)构建如上所述的dna展示模板包被珠,并进行下一选择轮次。
在一个实施方案中,标记的靶蛋白是荧光标记的靶蛋白。
在一个实施方案中,所述选择是facs选择。
在一个方面,本文提供了提供rna编码dna展示文库的方法,其包括:(i)将氨基烯丙基三磷酸核苷(aa-ntp)与n-羟基琥珀酰亚胺(nhs)-peg-马来酰亚胺(nhs-peg)缀合,形成ntp-peg-马来酰亚胺(ntp-peg);(ii)提供用还原硫醇官能化的dna多核苷酸(硫醇-dna)群体;(iii)将用还原硫醇官能化的dna多核苷酸(硫醇-dna)与ntp-peg上的马来酰亚胺缀合,形成ntp-peg-dna;和(iv)纯化ntp-peg-dna缀合物。
在一个方面,本文提供了提供rna编码dna展示文库的方法,包括:(i)将氨基烯丙基三磷酸核苷(aa-ntp)与n-羟基琥珀酰亚胺(nhs)-peg-马来酰亚胺(nhs-peg)缀合,形成ntp-peg-马来酰亚胺(ntp-peg);(ii)提供包含用还原硫醇官能化的dna多核苷酸(硫醇-dna)的dna文库;(iii)将用还原硫醇官能化的dna多核苷酸(硫醇-dna)与ntp-peg上的马来酰亚胺缀合,形成ntp-peg-dna;和(iv)纯化ntp-peg-dna缀合物。
在一个方面,本文提供了制备蛋白质文库的方法,包括:(i)提供dna展示编码链的群体,其各自包含正向引物位点、t7rna聚合酶启动子、翻译起始密码子、编码区和反向引物结合位点;(ii)提供正向引物缀合珠;(iii)将每个dna展示编码链退火至正向引物缀合珠;(iv)在dna聚合酶存在下通过pcr将正向引物缀合珠上的引物延伸以形成包含双链dna展示模板的珠;(v)使双链dna展示模板变性以形成包含单链dna展示模板链的珠;(vi)将包含反向引物的ntp-peg-dna缀合物与单链dna展示模板链退火;(vii)在dna聚合酶存在下通过pcr延伸ntp-peg-dna缀合物的反向引物以形成包含具有peg-ntp的双链dna展示模板的珠;(viii)在包含双链dna展示模板的珠上体外转录dna展示模板,产生包含dna部分和rna部分的dna-rna融合物;(ix)体外翻译rna部分以产生rna-蛋白质融合物群体,从而产生蛋白质文库。
在一个实施方案中,所述rna-蛋白质融合物包含通过嘌呤霉素连键与蛋白质连接的mrna。
在一个实施方案中,所述正向引物缀合珠由羧酸官能化珠与胺官能化正向引物之间形成的氨基键形成。
在一个方面,本文提供了选择靶蛋白的方法,包括:(i)提供包含正向引物位点、t7rna聚合酶启动子、翻译起始密码子、随机文库区和反向引物结合位点的dna编码链;(ii)提供正向引物缀合珠(羧酸官能化珠与胺官能化正向引物之间形成的氨基键);(iii)将dna展示文库编码链退火至正向引物缀合珠;(iv)在dna聚合酶存在下通过pcr将正向引物缀合珠上的引物延伸以形成包含双链dna展示模板的珠;(v)使双链dna展示模板变性以形成包含单链dna展示模板链的珠;(vi)将包含反向引物的ntp-peg-dna缀合物与单链dna展示模板链退火;(vii)在dna聚合酶存在下通过pcr将ntp-peg-dna缀合物的反向引物延伸,形成包含具有peg-ntp的双链dna展示模板的珠;(viii)在包含双链dna展示模板的珠上体外转录dna展示模板,产生包含dna部分和rna部分的dna-rna融合物;(ix)体外翻译rna部分以产生rna-蛋白质融合物群体,从而产生蛋白质文库;和(x)基于融合结合或活性选择所需的rna-蛋白质融合物,从而选择所述所需蛋白质和编码所述蛋白质的所述核酸。
在一个实施方案中,所需蛋白质是抗体、抗体片段、适体酶和酶。
在一个实施方案中,通过facs选择所需蛋白质。
在一个实施方案中,三磷酸核苷(aa-ntp)是三磷酸腺苷(atp)、三磷酸鸟苷(gtp)、三磷酸胞苷(ctp)、三磷酸5-甲基尿苷(m5utp)、三磷酸尿苷(utp)、任何非天然三磷酸核苷。
在一个实施方案中,rna聚合酶底物是为附着于peg而添加的替代连接化学物质。
在一个实施方案中,所述接头是peg。
在一个实施方案中,peg是peg3100、peg3200、peg3300、peg3400、peg3500或peg3600。
在一个实施方案中,所述接头不包含多核苷酸。
在一个实施方案中,接头的长度为100-200埃、200-250埃、250-280埃、280-300埃、300-400埃。
在一个方面,本文描述了包含脱氧核糖多核苷酸、接头分子和三磷酸核苷的dna-rna融合物。
在一个方面,本文描述了双链dna-rna融合物,其包含与dna展示编码链互补的dna展示模板链,只是所述dna展示编码链在其5'末端还包含接头分子和rna分子,并且其中所述rna分子从dna展示编码链转录。
在一个实施方案中,所述接头分子是聚乙二醇。
在一个方面,本文提供了将转录产物捕获到支架的方法。
在一个实施方案中,所述支架是固体支持物。
在一个实施方案中,所述固体支持物是珠、膜或过滤器。
在某些实施方案中,dna编码链、dna编码序列、候选dna序列的群体包括至少109、优选至少1010、更优选至少1011、1012、或1013,和最优选至少1014个不同的rna;使用含有atp、ctp、gtp、utp、dtt、rna酶抑制剂、t7rna聚合酶和dna展示模板的缓冲溶液进行体外转录反应;选择步骤包括将所需的rna与固定的结合配偶体结合;选择步骤包括测定所需rna的功能活性;dna分子被扩增;该方法还包括重复上述选择方法的步骤;该方法还包括从dna分子转录rna分子并重复该循环。
一方面,本文提供了通过富集序列库来选择所需rna或所需dna的方法。该方法包括以下步骤:(a)提供候选dna分子群体,其各自包含正向引物位点、t7rna聚合酶启动子、dna编码区和反向引物结合位点;(ii)将每个dna编码链退火至正向引物;(iii)在dna聚合酶存在下延伸引物以形成双链dna展示模板群体;(iv)使双链dna展示模板群体变性以形成单链dna展示模板链群体;(v)将包含反向引物的ntp-接头-dna缀合物退火至每个单链dna展示模板链;(vi)在dna存在下通过pcr延伸ntp-接头-dna缀合物的反向引物以形成包含接头-ntp的双链dna展示模板群体;(vii)体外或原位转录候选dna编码序列以产生候选dna-rna融合物群体;(viii)在使结合配偶体-dna-rna融合物复合物与未结合的群体成员基本分开的条件下,使dna-rna融合物群体与dna-rna融合物的dna部分或rna部分的特异性结合配偶体接触;(ix)从复合物中释放结合的dna-rna融合物;和(x)在使结合配偶体-dna-rna融合物复合物与所述未结合的群体成员基本分开的条件下,将步骤(ix)的dna-rna融合物群体与所需dna-rna融合物的rna部分的特异性结合配偶体接触,从而选择所需的rna。
在某些实施方案中,该方法还包括重复步骤(i)至(x)。此外,对于这些重复步骤,可以以任何顺序使用相同或不同的结合配偶体来选择性富集所需的dna-rna融合物。在另一个实施方案中,步骤(x)包括使用对所需融合物的rna部分特异的结合配偶体。该步骤在融合物的rna部分的逆转录之后进行,以产生编码所需蛋白质的dna。如果需要,可以分离和/或pcr扩增该dna。
在相关方面,本文提供了制备文库(例如蛋白质,dna-rna文库,rna适体文库)的方法或选择所需分子(例如蛋白质,dna或rna分子或具有特定功能或改变功能的分子)的方法。
在另一相关方面,本文提供了用于实施本文所述的任何选择方法的试剂盒。
一方面,本文提供了包含固定化单链核酸阵列的微芯片,所述核酸与dna-rna融合物杂交。优选地,dna-rna融合物的rna组分由dna编码。
4.附图说明
图1用于产生ntp-peg-dna缀合物的化学连接反应示意图。在反应1中,氨基烯丙基三磷酸核苷(aa-ntp)与异双官能peg上的n-羟基琥珀酰亚胺(nhs)缀合。这产生连同nhs离去基团一起的缀合物,在反应2之前使用尺寸排阻色谱法除去连同过量的aa-ntp一起的nhs离去基团。在反应2中,用还原硫醇官能化的dna寡核苷酸与反应1的官能peg-ntp缀合物上的马来酰亚胺缀合。在该反应之后,使用阴离子交换色谱来纯化最终的ntp-peg-dna产物。
图2构建dna展示模板包被珠。a)dna展示文库编码链设计。dna展示文库编码链包含正向引物位点、t7rna聚合酶启动子、随机文库区和反向引物结合位点。b)蛋白编码dna展示文库编码链设计。蛋白编码dna展示文库编码链包含正向引物位点、t7rna聚合酶启动子、aug起始密码子、随机文库区和反向引物结合位点。c)正向引物缀合珠。正向引物与珠缀合可以是在羧酸官能化珠与胺官能化正向引物之间形成的氨基键。d)通过dna聚合酶的dna展示模板链延伸。dna展示编码链退火至fp珠,pcr的重复循环允许dna聚合酶形成用双链dna展示模板装饰的珠。e)双链dna展示模板偶联到在pcr之后形成的珠上。f)用单链dna展示模板链装饰的珠。为了实现用单链dna展示模板链装饰的珠,将偶联到珠的双链dna展示模板变性。g)使用ntp-peg-dna反向引物通过dna聚合酶延伸dna展示编码链。dna展示模板链退火至ntp-peg-dna反向引物,pcr的重复循环允许dna聚合酶形成用具有通过peg接头连接的ntp的双链dna展示模板装饰的珠。h)具有通过peg接头连接的ntp的完整的珠偶联双链dna展示模板。
图3dna展示示意图。a)具有通过peg接头连接的ntp的完整的双链dna展示模板。b)转录dna展示模板,其中缀合的ntp插入到新形成的rna链中。c)dna模板上展示的rna。d)dna展示模板进行转录以产生在dna上展示的rna。在dna模板转录后,通过rna聚合酶将缀合三磷酸核苷插入到新形成的rna中,产生模板dna与其编码的rna之间的共价连接。通过dna展示模板展示rna的dna展示可用于在转录后捕获rna。
图4证明dna展示的聚丙烯酰胺凝胶电泳。泳道1为10bpdna梯。泳道2是rna梯。泳道3显示具有转录的dna展示模板。泳道4显示没有转录的dna展示模板。泳道5显示具有转录和rna酶处理的dna展示模板。泳道6显示dna酶处理后来自珠的dna的损失。泳道7显示dna酶处理后的穿流。泳道8是转录后的游离rna。当比较泳道3,具有转录的dna展示模板,与泳道4,没有转录的dna展示模板时,可以在带移位中看到由于转录的rna的连接而导致的质量增加。泳道5,具有转录然后进行rna酶处理的dna展示模板,证实了这种移位是由于rna,因为当rna酶降解所连接的rna时,条带移位回到原始的仅dna的位置。
图5各种展示的示意图。1.粒子展示;2.粒子展示和dna展示;3.粒子展示、dna展示和mrna展示。
4.1定义
如本文所用,“群体”是指超过一个分子(例如超过一个rna,dna或dna-rna融合分子)。因为所公开的方法有利于以大量候选分子起始(如果需要的话)的选择,“群体”优选表示超过109个分子,更优选超过1011、1012或1013个分子,并且最优选超过1013个分子。
“选择”是指使群体中的一个分子与其它分子基本分隔。如本文所用,“选择”步骤相对于在选择步骤之后群体中的不期望分子提供至少2倍、优选30倍、更优选100倍、最优选1000倍富集所需分子。如本文所示,选择步骤可以重复任意次数,并且可以在给定方法中组合不同类型的选择步骤。
“蛋白质”是指任何两个或更多个通过一个或多个肽键连接的天然存在或修饰的氨基酸。本文中可互换使用“蛋白质”和“肽”。
“rna”是指两个或更多个共价键合的天然存在或修饰的核糖核苷酸的序列。
“起始密码子”是指发出蛋白编码序列开始的信号的三个碱基。一般来说,这些碱基是aug(或atg);然而,可以用能够以这种方式利用的任何其它碱基三联体代替。
“共价键合”是指直接通过共价键或间接通过另一个共价键合的序列。
“改变的功能”是指分子功能的任何定性或定量的变化。
本文所用的“结合配偶体”是指对所需dna-rna融合物的一部分具有特异性、共价或非共价亲和力的任何分子。结合配偶体的实例包括但不限于以下成员:抗原/抗体对、蛋白/抑制剂对、受体/配体对(例如细胞表面受体/配体对,例如激素受体/肽激素对)、酶/底物对(例如激酶/底物对)、凝集素/碳水化合物对、寡聚或异寡聚蛋白质聚集体、dna结合蛋白/dna结合位点对、rna/蛋白质对和核酸双链体、异源双链体或连接的链、以及能够与dna-rna融合物的任何部分形成一个或多个共价或非共价键(例如二硫键)的任何分子。
“固体支持物”是指但不限于任何柱(或柱材料)、珠、试管、微量滴定盘、固体颗粒(例如琼脂糖或琼脂糖凝胶)、微芯片(例如硅、硅玻璃或金芯片)、或可直接或间接(例如通过其它结合配偶体中间体、例如其它抗体或蛋白质a)结合亲和复合物、或其中可以嵌入亲和复合物(例如通过受体或通道)的膜(例如脂质体或囊泡的膜)。
5.发明详述
特异性结合其它分子的分子对于许多生物医学和分析应用是必不可少的,例如治疗学、诊断学、实验室研究和分析科学的许多方面。本公开提供了许多显著的优点。本公开允许使用具有相当长度的候选分子群体进行重复轮次选择。本公开涉及用于生物医学和分析应用的结合配体的发现和进化的dna展示的新方法。这些结合配体包括但不限于核酸或蛋白质(包括但不限于抗体、抗体片段、肽、多肽)。除了选择结合配体之外,dna展示还用于选择和改善催化活性,包括但不限于酶、核酶和适体酶。
公开的dna展示是在结合配体回到编码该分子的dna之间进行连接的过程。本文公开了dna展示模板由三个部分构成:dna模板、接头和ntp,其是该过程的衔接子。该连接允许选择结合混合的结合分子群体中的所需靶的特定分子。然后编码结合分子的dna可用于产生结合分子或产生新的结合分子。dna-rna融合物提供了选择可用于药物和生物技术的结合分子的改进方法。
另一个优点是,本发明的选择和定向进化技术可以利用非常大且复杂的候选序列文库。大的文库大小为定向进化应用提供了优势。候选库大小可能是约1015个候选物。随机区可以分离或在所需dna-rna融合物的背景中进行筛选。大多数(如果不是全部)可能的序列可在dna-rna融合物的候选库中表达。产生靶标的结合分子涉及重复的选择循环。将结合分子的混合库暴露于靶标,将弱结合物洗去并且保持更强的结合物。然后将这些更强的结合物一起增殖或扩增,然后组成新的结合分子库,用于下一个结合分子进化循环。重复该循环,增加分子库的结合能力,直到获得非常强的结合分子。对于珠选择技术上的适体,在转录后有空间信息丢失。使用本文公开的本发明的dna展示可以避免该信息丢失。
本文描述了使用其中rna与其编码dna共价连接的融合物从具有期望功能的rna群体中选择rna的通用方法。通过体外或原位延伸和转录包含正向引物区、t7rna聚合酶启动子、dna编码区和反向引物区的dna库合成这些dna-rna融合物(图2a)。rna与dna之间的共价连接(以ntp-接头-dna缀合物的形式)允许dna的空间信息与其编码的rna的空间信息内在联系。这对于在珠选择技术例如facs上的适体是至关重要的。
5.1ntp-接头-dna的合成
为了产生dna-rna融合物的模板,制备了ntp-接头-dna缀合物。氨基烯丙基三磷酸核苷(aa-ntp)与接头缀合。在一个实施方案中,接头是异双官能peg。aa-ntp与异双官能peg上的n-羟基琥珀酰亚胺(nhs)缀合。这导致具有nhs离去基团的缀合物,其中使用尺寸排阻色谱法除去过量的aa-ntp(图1,反应1)。用还原硫醇官能化的dna寡核苷酸与来自反应1的官能peg-ntp缀合物上的马来酰亚胺缀合。在该反应之后,使用阴离子交换色谱来纯化最终的ntp-peg-dna缀合物。
在一个实施方案中,氨基烯丙基三磷酸胞苷(aa-ctp)通过nhs酯与异双官能nhs-peg-马来酰亚胺接头缀合,形成如图1所示的酰胺键。然后将ctp-peg-马来酰亚胺缀合物与还原的5'c6s-s硫醇修饰的dna寡核苷酸反应以形成ctp-peg-dna,如图1所示。在一个实施方案中,dna寡核苷酸是图2的反向引物。
在其它实施方案中,三磷酸核苷是三磷酸腺苷(atp)、三磷酸鸟苷(gtp)、三磷酸胞苷(ctp)、三磷酸5-甲基尿苷(m5utp)、三磷酸尿苷(utp)、任何非天然三磷酸核苷或具有为附着于peg而添加的任何替代连接化学物质的任何rna聚合酶底物。
在某些实施方案中,与硫醇-马来酰亚胺连接化学物质一起使用peg接头。除了peg之外的接头可以与一系列连接化学物质一起使用。
使用的peg接头的长度是重要的,因为太短将不允许缀合的ntp插入到新形成的rna链中,而太长可能导致早期插入或可能增加非特异性ntp插入的可能性。在某些实施方案中,ntp-接头-dna缀合物包含长度为200-300埃的接头。在某些实施方案中,接头的长度为280埃。
在某些实施方案中,接头不包含多核苷酸。
在某些实施方案中,peg是peg3100、peg3200、peg3300、peg3400、peg3500或peg3600。
在某些实施方案中,peg是peg3400。
5.2构建dna展示模板包被珠
图2显示了本公开的dna展示方案的一个实施方案。涉及构建dna展示模板包被珠的步骤通常如下进行:
(a)dna展示编码链设计(图2a)。用于典型的rna编码dna展示文库的dna展示文库编码链设计如图2a所示。dna展示编码链包含正向引物位点、t7rna聚合酶启动子、多达但不限于30kb的随机dna编码区和反向引物结合位点。在其它实施方案中,dna编码区至少为,但不限于10-15kb、15-20kb、20-25kb、25-30kb、30-35kb、35-40kb、40-45kb、45-50kb。
在一个实施方案中,为了使用流式细胞术进行选择,将编码链文库扩增到珠上。
(b)正向引物缀合珠(图2c)。正向引物通过羧酸官能化珠与胺官能化正向引物之间形成的氨基键与珠缀合。
(c)通过dna聚合酶延伸dna展示模板链(图2d)。为了扩增dna展示编码链,通过dna聚合酶延伸与dna展示编码链互补的dna展示模板链(图2a)。将dna展示编码链退火至fp珠,并使用乳液pcr进行扩增,该方式使得每个珠展示单个dna展示编码链的许多拷贝,与wang等人相似。(wang等人,2014)(图2d)。
(d)在pcr之后形成偶联于珠的双链dna展示模板(图2e)。
(e)用单链dna展示模板链装饰的珠。乳液破坏后,珠上的双链dna(图2e)变性为单链dna展示模板链(图2f)。
(f)使用ntp-peg-dna反向引物通过dna聚合酶延伸dna展示编码链。dna为反向引物的ntp-peg-dna退火至单链dna编码链,并在dna聚合酶存在下通过pcr扩增(图2g)。
(g)具有通过peg接头连接的ntp的完整的珠偶联双链dna展示模板(图2h)。
在另一个实施方案中,如上所述构建dna展示,但不包括珠。
5.3dna展示模板的构建
dna展示模板的构建包括的步骤通常如下进行:
(a)dna展示编码链设计。用于典型的rna编码dna展示文库的dna展示文库编码链设计如图2a所示。dna展示编码链包含正向引物位点、t7rna聚合酶启动子、随机dna编码区和反向引物结合位点。
在一个实施方案中,dna展示与珠不相关。
(b)通过dna聚合酶延伸dna展示模板链。为了扩增dna展示编码链,通过dna聚合酶延伸与dna展示编码链互补的dna展示模板链。使用pcr扩增dna展示编码链。
(c)在pcr之后形成双链dna展示模板。
(d)乳液破坏后,双链dna变性为单链dna展示模板链。
(e)使用ntp-peg-dna反向引物通过dna聚合酶延伸dna展示编码链。将dna是反向引物的ntp-peg-dna退火至单链dna编码链,并在dna聚合酶存在下通过pcr扩增。
(f)使用ntp-peg-dna反向引物通过dna聚合酶延伸dna展示编码链。将dna是反向引物的ntp-peg-dna退火至单链dna编码链,并在dna聚合酶存在下通过pcr扩增。
(g)具有通过peg接头连接的ntp的双链dna展示模板。
5.4展示蛋白质编码rna的dna展示模板包被珠的构建
构建蛋白质编码dna展示模板包被珠包括的步骤通常如下进行:
(a)蛋白质编码dna展示编码链设计(图2b)。用于在dna展示文库中展示蛋白质编码rna的dna展示文库编码链设计如图2b所示。蛋白质编码dna展示编码链包含正向引物位点、t7rna聚合酶启动子、起始密码子、随机dna编码区和反向引物结合位点。
在一个实施方案中,为了使用流式细胞术选择,将编码链文库扩增到珠上。
(b)正向引物缀合珠。正向引物通过羧酸官能化珠与胺官能化正向引物之间形成的氨基键与珠缀合。
(c)通过dna聚合酶延伸dna展示模板链。为了扩增dna展示编码链,通过dna聚合酶延伸与dna展示编码链互补的dna展示模板链(图2b)。将dna展示编码链退火至fp珠,并使用乳液pcr进行扩增,该方式使得每个珠展示单个dna展示编码链的许多拷贝,与wang等人相似。(wang等人,2014)(图2d)。
(d)在pcr之后形成偶联于珠的双链dna展示模板。
(e)用单链dna展示模板链装饰的珠。乳液破坏后,珠上的双链dna(图2e)变性为单链dna展示模板链。
(f)使用ntp-peg-dna反向引物通过dna聚合酶延伸dna展示编码链。dna为反向引物的ntp-peg-dna退火至单链dna编码链,并在dna聚合酶存在下通过pcr扩增。
(g)具有通过peg接头连接的ntp的完整的珠偶联双链dna展示模板。
在另一个实施方案中,如上所述构建dna展示,但不包括珠。
5.5展示蛋白质编码rna的dna展示模板的构建
dna展示模板的构建包括的步骤通常如下进行:
(a)蛋白质编码dna展示编码链设计(图2b)。用于在dna展示文库中展示蛋白质编码rna的dna展示文库编码链设计如图2b所示。蛋白质编码dna展示编码链包含正向引物位点、t7rna聚合酶启动子、起始密码子、随机dna编码区和反向引物结合位点。
(b)通过dna聚合酶延伸dna展示模板链。为了扩增dna展示编码链,通过dna聚合酶延伸与dna展示编码链互补的dna展示模板链。使用pcr扩增dna展示编码链。
(c)在pcr之后形成双链dna展示模板。
(d)乳液破坏后,双链dna变性为单链dna展示模板链。
(e)使用ntp-peg-dna反向引物通过dna聚合酶延伸dna展示编码链。将dna为反向引物的ntp-peg-dna退火至单链dna编码链,并在dna聚合酶存在下通过pcr扩增。
(f)使用ntp-peg-dna反向引物通过dna聚合酶延伸dna展示编码链。将dna为反向引物的ntp-peg-dna退火至单链dna编码链,并在dna聚合酶存在下通过pcr扩增。
(g)具有通过peg接头连接的ntp的双链dna展示模板。
5.7构建dna展示模板包被珠
代替将ntp-peg与dna寡核苷酸引物连接,ntp-peg可以直接缀合到文库dna。这意味着每轮选择需要更少轮的链延伸,但是每一轮必须包括缀合步骤。
在一个实施方案中,用于典型的rna编码dna展示文库的dna展示文库编码链设计包含正向引物位点、t7rna聚合酶启动子、与ntp-接头直接连接的随机dna编码区。在一个实施方案中,接头是peg。然后在dna展示模板上进行体外转录,并且peg接头缀合的ntp掺入形成的rna链。这导致rna展示在dna模板上。dna展示可以与珠相关或与珠无关。在一个实施方案中,dna编码模板链包含起始密码子。在一个实施方案中,dna编码模板链不包含起始密码子。在一个实施方案中,起始密码子是aug。
5.8在dna展示模板上展示rna
然后在dna展示模板包被珠上进行体外转录,并且peg接头缀合的ntp掺入形成的rna链中(图3)。这导致rna展示在dna模板上。使用该方法,在一个实施方案中,可以制备rna适体的文库,其然后可以进行选择。
5.9选择和扩增
然后将珠上的rna适体与荧光靶蛋白质孵育并洗涤以准备用于facs选择。然后选择的rna适体可以使用粒子pcr扩增其dna模板(wang等人,2014)。如果已经进行了充分的选择轮次,则可以测序该文库以鉴定分离的适体,否则该文库可用于构建下一代dna展示模板包被珠用于下一轮选择。
在珠上选择蛋白质的情况下,如上所述,使用乳液pcr将蛋白质编码dna展示文库(图2b)附着于珠上。然后可以进行体外转录以产生珠上的rna文库。然后可以在rna展示珠上进行mrna展示技术(us6261804b1)(wilson,keefe等人2001)以产生蛋白展示珠。这通过连接到珠上的dna展示模板来有效地在rna上展示蛋白质。然后可以对珠上的蛋白进行facs选择。
5.10dna模板的制备
作为旨在产生dna-rna融合物的步骤,合成融合物的dna部分。这可以通过直接化学dna合成来实现,或者更常见的是通过在dna聚合酶存在下使用pcr延伸dna并纯化任何双链dna模板来实现。
这样的dna模板可以通过任何标准技术(包括重组dna技术、化学合成或两者的任何技术)来产生。原则上,可以使用允许生产包含已知、随机、随机化或诱变的序列的一个或多个模板的任何方法用于该目的。在一个特定的方法中,合成寡核苷酸(例如,含有随机碱基),并在转录前将其扩增(例如通过pcr)。化学合成也可用于产生随机盒,然后将其插入已知蛋白质编码序列的中间(参见例如第8.2章,ausubel等人,currentprotocolsinmolecularbiology,johnwiley&sonsandgreenepublishingcompany,1994)。
dna模板序列的总随机化的替代方案是部分随机化,并且以这种方式合成的库通常被称为“掺杂”库。对rna序列进行的这种技术的一个实例描述于例如ekland等人(nucl.acidsresearch23:3231(1995))。部分随机化可以通过偏置合成反应而化学地进行,使得每个碱基加成反应混合物含有过量的一种碱基和少量的各其它碱基;通过仔细控制碱基浓度,通过这种方法可以实现需要的突变频率。也可以例如beaudry和joyce(science257:635(1992))和bartel和szostak(science261:1411(1993))中所述的,使用易错pcr技术来产生部分随机化的库。
许多方法也可用于产生以已知序列开始的dna构建体,并且如果需要,则产生诱变的dna库。这种技术的实例描述于ausubel等人(同上,第8章);sambrook等人(molecularcloning:alaboratorymanual,第15章,coldspringharborpress,newyork,2.sup.nded.(1989);cadwell等人(pcrmethodsandapplications2:28(1992));tsang等人(meth.enzymol.267:410(1996));reidhaar-olsen等人(meth.enzymol.208:564(1991));以及ekland和bartel(nucl.acids.res.23:3231(1995))。随机序列也可以通过stemmer(nature370:389(1994))中概述的“改组”技术产生。最后,一组两个或更多个同源基因可以在体外重组以产生起始文库(crameri等人.nature391:288-291(1998))。
5.11rna的产生
如上所述,通过体外转录dna模板产生rna部分。在一个优选的方法中,t7聚合酶用于酶促产生rna链。转录通常以与pcr反应相同的体积进行(来自100μml反应的pcrdna用于100μml的转录)。用于该用途的其它合适的rna聚合酶包括但不限于sp6、t3和大肠杆菌rna聚合酶(描述于例如ausubel等人(同上,第3章)。另外,合成的rna可以是全部或者部分修饰的rna。在一个具体实例中,可以使用修饰的核糖核苷酸和标准技术产生硫代磷酸酯rna(例如,通过t7转录)。这种修饰的rna提供了核酸酶稳定的优点。然后如前所述,使用脲page,然后在nap-25(pharmacia)上脱盐,从转录反应纯化全长rna样品(roberts和szostak,proc.natl.acad.sci.usa94:12297-12302(1997))。
5.12dna-rna融合物的产生和恢复
为了产生dna-rna融合物,可以使用任何体外或原位转录系统。然而,原则上,允许形成dna-rna融合物并且不显著降解融合物的rna部分的任何转录系统可用于本发明。此外,为了减少任何这些系统中的rna降解,降解阻断反义寡核苷酸可以包括在转录反应混合物中;这样的寡核苷酸特异性杂交并覆盖触发降解的分子的rna部分内的序列。
在一个实施方案中,任何数量的真核转录系统可用于使用。
一旦产生,可以通过dna或rna纯化的任何标准技术从转录反应混合物中回收dna-rna融合物。纯化也可以基于融合物的rna部分;例如,在ausubel等人(同上,第4章)中描述了用于这种纯化的技术。
5.12选择所需的dna-rna融合物
可以通过任何可用于选择性地从候选融合物群体分隔或分离所需融合物的手段来实现所需dna-rna融合物的选择。分离技术的实例包括但不限于催化活性、活性测定中的特定效果、选择性结合、结合特异性,例如对于直接或间接固定在柱、珠、膜或其它固体支持物上的结合配偶体。这些技术中的第一种利用固定化的选择基序,其可以由可能结合的任何类型的分子组成。选择也可以基于连接到与候选分子反应的亲和标记(例如底物-生物素)的底物分子的使用,或基于与融合分子的任何其它类型的相互作用。此外,可以基于其催化活性来选择蛋白质;根据该特定技术,基于其将靶分子连接到其自身的能力来选择所需分子,然后基于该靶标的存在分离功能分子。通过本公开能够实现使用相同方法或任何其它功能选择来分离新颖或改进的适体的选择方案。
此外,如果重复靶向融合物的相同部分(例如rna部分)的富集步骤,则优选使用不同的结合配偶体。在本文描述的一个具体实例中,通过首先使用对融合物的dna部分特异的结合配偶体,然后在两个连续的步骤中,使用两个不同的结合配偶体(两者对融合物的rna部分是特异性的)而富集分子群体的所需融合物。同样,这些复合物可以通过任何标准分离技术从样品组分分离,包括但不限于柱亲和色谱或离心。
可以根据任何标准方案进行具有所需活性(例如催化活性、结合亲和力、结合特异性或活性测定中的特定效果)的反应产物的选择和/或筛选。由于可以通过pcr扩增微量的dna,因此这些选择可以以这种规模进行,从而能够真正广泛地搜寻期望的活性,既经济又有效。
可以选择或分隔展示文库以结合靶分子。在这种情况下,选择或分隔是指结合靶分子的文库成员与未结合靶分子的文库成员分离的任何过程。选择可以通过本领域已知的各种方法来实现。在大多数应用中,优选与靶分子的结合是选择性的,使得相对于其它结合事件偏爱与靶标的结合。最终,使用本发明鉴定的结合分子可用作治疗试剂和/或诊断剂。
可以执行选择策略以允许对几乎任何靶标进行选择。重要的是,选择策略不需要关于靶分子或关于展示文库成员的任何详细结构信息。整个过程由结合到给定靶分子的文库成员所涉及的结合亲和力和特异性驱动。
使用标准分子生物学,通过其编码核酸可以容易地鉴定选定的文库成员。本公开广泛地允许鉴定任何已知靶分子的结合分子。另外,通过分离所选择的文库成员的结合分子,可以发现新的未知靶标,并将其用于靶分子的鉴定和验证。
来自展示文库的结合分子的选择可以以任何形式进行,以鉴定结合文库成员。结合选择通常包括固定期望的靶分子,添加展示文库,允许结合,以及通过洗涤去除非结合物/弱结合物。保持与靶标结合的富集文库可以用例如酸、离液盐、加热、用已知配体的竞争性洗脱、高盐、碱、靶标的蛋白水解性释放、核酸的酶促释放洗脱。在某些实施方案中,使用相同或更严格的条件或使用不同的结合形式对洗脱的文库成员进行更多轮的结合和洗脱,这将增加富集。在其它实施方案中,结合文库成员未从靶标洗脱。为了选择与细胞表面上表达的蛋白质(例如离子通道或跨膜受体)结合的文库成员,细胞本身可以用作选择剂。选择程序还可以涉及选择与细胞表面受体的结合,所述细胞表面受体经内化,使得受体与结合分子一起进入细胞质、细胞核或其它细胞区室,例如高尔基体或溶酶体。所述区室的隔离导致被内化的文库成员与非内化的文库成员分隔(hart等人,jbiolchem,269,12468-74,1994)。选择程序还可以涉及体内选择。富集的文库的核酸部分可以通过例如pcr扩增,导致许多级次的扩增,允许通过例如,克隆和dna测序鉴定。
根据亲和选择的具体实施方案,由特定成员产生的反应产物的文库在结合条件下与靶标接触。如果一种或多种所形成的化合物对靶标具有亲和力,则将导致结合。在随后的步骤中,分隔结合文库成员或其衍生的核酸。附着于形成的化合物的核酸随后通过例如,pcr扩增以产生核酸的多个拷贝,其编码显示所需亲和力的化合物的合成历史。扩增的核酸可以通过许多众所周知的技术进行测序,以解码参与成功化合物形成的化学基团。或者,扩增的核酸可用于形成下一代文库。
5.13dna展示与mrna展示的组合
如上所述,形成模板的dna允许展示rna,在一个实施方案中,rna是rna适体。如上所述,rna适体文库可以展示在珠上。跨接粒子展示与rna展示之间的间隙意味着可以在珠粒上选择蛋白质,例如抗体或酶,在没有本发明公开的dna展示技术的情况下这是不可能的。这在图5中示出。这允许rna适体的荧光激活细胞分选(facs),可能产生比先前的rna适体选择方法强1000倍的结合亲和力。
如本文所公开的dna展示连接两种其它技术:粒子展示和mrna展示,如美国专利号6,261,804中所公开的。mrna展示由体外或原位转录/翻译方案组成,其产生与其自身的mrna的3'末端共价连接的蛋白质,即rna-蛋白质融合物。这通过mrna分子与附着于其3'末端的肽受体的合成和体外或原位翻译来实现。一种优选的肽受体是嘌呤霉素,一种核苷类似物,其添加到生长中的肽链的c-末端并终止翻译。在一个优选的设计中,dna序列包括在信使的末端与肽受体之间,肽受体被设计成使得核糖体在开放阅读框的末端停留,为肽受体(例如嘌呤霉素)提供额外的时间以在水解肽基-trna连键前接受新生肽链。
如果需要,所得到的rna-蛋白质融合物允许重复轮次的选择和扩增,因为可以通过逆转录和扩增来回收蛋白质序列信息(例如,通过pcr扩增以及任何其它扩增技术,包括基于rna的扩增技术,例如3sr或tsa)。然后可以将扩增的核酸转录、修饰和体外或原位翻译以产生用于下一轮选择的mrna-蛋白质融合物。进行多轮选择和扩增的能力使得能够富集和分离非常罕见的分子,例如1015个成员库中的一个所需分子。这继而允许分离特异性识别几乎任何靶标或催化所需化学反应的新的或改进的蛋白质。
因此,在一个实施方案中,本文提供了粒子展示-dna展示-mrna展示系统。本文提供了一种制备蛋白质文库的方法,其包括:(i)提供dna展示编码链群体,其各自包含正向引物位点、t7rna聚合酶启动子、翻译起始密码子、编码区和反向引物结合位点;(ii)提供正向引物缀合珠;(iii)将每个dna展示编码链退火至正向引物缀合珠;(iv)在dna聚合酶存在下通过pcr延伸正向引物缀合珠上的引物以形成包含双链dna展示模板的珠;(v)使双链dna展示模板变性以形成包含单链dna展示模板链的珠;(vi)将包含反向引物的ntp-peg-dna缀合物与单链dna展示模板链退火;(vii)在dna聚合酶存在下通过pcr延伸ntp-peg-dna缀合物的反向引物以形成包含具有peg-ntp的双链dna展示模板的珠;(viii)在包含双链dna展示模板的珠上体外转录dna展示模板,产生包含dna部分和rna部分的dna-rna融合物;和(ix)体外翻译rna部分以产生rna-蛋白质融合物群体。与rna展示结合的dna展示的产物是dna-rna-蛋白质融合物(图5,子图3)。
在一个实施方案中,rna-蛋白质融合物包含通过嘌呤霉素连键而连接至蛋白质的mrna。
在一个实施方案中,正向引物缀合珠通过在羧酸官能化珠与胺官能化正向引物之间形成的氨基键形成。
本文还提供了选择靶蛋白的方法,其包括:(i)提供包含正向引物位点、t7rna聚合酶启动子、翻译起始密码子、随机文库区和反向引物结合位点的dna编码链;(ii)提供正向引物缀合珠(羧酸官能化珠与胺官能化正向引物之间形成的氨基键);(iii)将dna展示文库编码链退火至正向引物缀合珠;(iv)在dna聚合酶存在下重复进行pcr循环,以将正向引物缀合珠上的引物延伸以形成包含双链dna展示模板的珠;(v)使双链dna展示模板变性以形成包含单链dna展示模板链的珠;(vi)将包含反向引物的ntp-peg-dna缀合物与单链dna展示模板链退火;(vii)在dna聚合酶存在下通过pcr将ntp-peg-dna缀合物的反向引物延伸,形成包含具有peg-ntp的双链dna展示模板的珠;(viii)在包含双链dna展示模板的珠上体外转录dna展示模板,产生包含dna部分和rna部分的dna-rna融合物;(ix)体外翻译rna部分以产生rna-蛋白质融合物群体,从而产生蛋白质文库;和(x)基于融合结合或活性选择所需的rna-蛋白质融合物,从而选择所述所需蛋白质和编码所述蛋白质的所述核酸。
在一个实施方案中,所需蛋白质是抗体、抗体片段、适体酶和酶。
在一个实施方案中,通过facs选择所需蛋白质。
在一个实施方案中,三磷酸核苷(aa-ntp)是三磷酸腺苷(atp)、三磷酸鸟苷(gtp)、三磷酸胞苷(ctp)、三磷酸5-甲基尿苷(m5utp)、三磷酸尿苷(utp)、任何非天然三磷酸核苷。
5.14使用dna展示的方法
通过dna展示模板的dna展示可用于在转录后捕获rna。这种捕获的应用可以是基于研究的,如转录产物的定量,或更多应用,如将基因型(dna)与表型(rna)相关联用于rna适体、dna适体、肽核酸(pna)和异种核酸(xna)的体外进化。本公开的选择系统在任何使用rna技术来解决治疗、诊断或工业问题的领域中具有商业应用。该选择技术可用于改进或改变现有rna以及分离具有所需功能的新适体。这些rna可以是天然存在的序列,可以是天然存在的序列的改变形式,或者可以是部分或完全合成的序列。此外,这些方法还可用于分离或鉴定有用的核酸或小分子靶标。
新型结合试剂的分离-在一个特定应用中,本文所述的dna-rna融合技术可用于分离具有特异性结合(例如配体结合)性质的dna适体、rna适体、肽核酸和异种核酸。显示高度特异性结合相互作用的适体可用作非抗体识别试剂,允许dna-rna融合技术绕过传统的单克隆抗体技术。通过该方法分离的抗体型试剂可用于任何使用传统抗体的领域,包括诊断和治疗应用。
核酸具有足够的能力形成各种二维和三维结构,和在其单体中可用的足够化学通用性以用作几乎任何化合物(无论是单体的还是聚合的)的配体(形成特异性结合对)。任何大小或组成的分子都可以作为靶标。用于产生目的靶标的适体的合适方法是selex方法,其涉及从候选寡核苷酸的混合物的选择和使用相同的一般选择方案的结合、分隔和扩增的逐步重复来实现几乎任何期望标准的结合亲和力和选择性。从优选包含一段随机化序列的核酸混合物开始,selex.tm方法包括以下步骤:在有利于结合的条件下将混合物与靶标接触,将未结合的核酸与特异性结合靶分子的核酸分隔,解离核酸-靶标复合物,扩增从核酸-靶标复合物解离的核酸以产生配体富集的核酸混合物,然后按需要通过尽可能多的循环重复结合、分隔、解离和扩增的步骤,以产生针对靶分子的高度特异性的高亲和力核酸配体。在某些实施方案中,pcr可以经历2-30个循环、2-10、10-20和20-30个循环。
已知存在多种核酸一级、二级和三级结构。已经显示最常涉及非沃森-克里克型相互作用的结构或基序被称为发夹环、对称和不对称凸起、假结(pseudoknot)和它们的无数组合。几乎所有已知的这种基序的实例都表明它们可以以不超过30个核苷酸的核酸序列形成。因此,通常优选的是用连续随机化区段的selex程序由含有约20-50个核苷酸的随机化区段的核酸序列引发。在一个实施方案中,dna编码区具有20-50个核苷酸。在一个实施方案中,dna编码区具有20-30个核苷酸。在一个实施方案中,dna编码区具有30-50个核苷酸。在一个实施方案中,dna编码区具有多于50个核苷酸。
新催化剂的分离-本公开也可用于选择新的催化适体。体外选择和进化可用于分离新型催化rna和dna。在该方法的一个具体实例中,可以通过选择与催化剂的过渡态的化学类似物结合来间接地分离催化剂。在另一个具体实例中,可以通过选择与底物共价键形成(例如,使用与亲和标签连接的底物)或通过切割(例如通过选择破坏特定键,从而从固体支持物释放文库的催化成员的能力)来进行直接分离。
5.14.1转录后捕获rna的方法
通过组装包含与ntp连接的模板dna的dna展示模板,产生所需的rna捕获条件。在dna展示模板翻译后,连接的ntp将被插入到新形成的rna链中,产生rna与编码它的dna之间的共价连接(图5b)。以这种方式可以实现rna捕获。
5.14.2转录产物的定量
基因型表型联系涉及在一些转换事件之后保留信息。该信息性质可以是遗传序列或空间上的,例如与珠的偶联连接。在dna模板上进行dna展示,得到的rnadna融合物可以进行mrna展示。在mrna展示中,将嘌呤霉素连接到rna链的末端,并且在翻译期间插入到新形成的多肽中。然后将该蛋白经由rna和编码它的dna偶联到珠上(图5c)。
量化基因被转录的程度构成了一个困难的科学问题。大多数方法涉及修饰基因或其环境至显著程度,例如插入报告转录物或将基因从其基因组中克隆出来用于表达。
补充所述基因末端的pcr引物可通过peg接头与ntp缀合。将此修饰的引物dna插入基因组后,在基因转录后,其将具有共价连接的其编码的rna。由于展示的rna的额外质量,位点特异性核酸酶消化和page上的条带分析将显示所述基因的质量位移。以这种方式,可以量化基因的转录。
5.15与mrna展示选择系统组合使用dna展示
本公开的选择系统在任何使用蛋白质技术来解决治疗、诊断或工业问题的领域中具有商业应用。该选择技术可用于改善或改变现有蛋白质以及用于分离具有所需功能的新蛋白质。这些蛋白质可以是天然存在的序列,可以是天然存在的序列的改变形式,或者可以是部分或完全合成的序列。此外,这些方法还可用于分离或鉴定有用的核酸或小分子靶标。
新型结合试剂的分离。在一个特定应用中,本文所述的dna-rna-蛋白质融合技术可用于分离具有特异性结合(例如配体结合)性质的蛋白质。显示高度特异性结合相互作用的蛋白质可用作非抗体识别试剂,允许dna-rna-蛋白质融合技术绕过传统的单克隆抗体技术。通过该方法分离的抗体型试剂可用于任何使用传统抗体的领域,包括诊断和治疗应用。
人抗体的改进。本公开还可以用于改善用于治疗多种疾病中的任何一种的人或人源化抗体。在该应用中,开发抗体文库并在体外筛选,消除了对诸如细胞融合或噬菌体展示等技术的需要。在一个重要的应用中,本发明可用于改进单链抗体文库(ward等人,nature341:544(1989);和goulot等人,j.mol.biol.213:617(1990))。对于该应用,可变区可以从人源构建(以尽可能减少接受者的可能的不利免疫反应),或者可以包含完全随机化盒(以最大化文库复杂性)。为了筛选改进的抗体分子,测试候选分子库与靶分子(例如,如图2所示固定的抗原)的结合。随着选择从一轮进行到下一轮,更高级别的严格性被应用于结合步骤。为了增加严格性,改变诸如洗涤步骤的数量、过量竞争剂的浓度、缓冲条件、结合反应时间的长度和固定化基质的选择等条件。
单链抗体可以直接用于治疗或间接用于标准抗体的设计。这些抗体具有许多潜在的应用,包括分离抗自身免疫抗体、免疫抑制、以及用于病毒性疾病例如aids的疫苗的开发。
新催化剂的分离。本公开还可以用于选择新的催化蛋白。以前已经使用体外选择和进化来分离新的催化性rna和dna,并且在本公开中,用于分离新的蛋白质酶。在该方法的一个具体实例中,可以通过选择与催化剂的过渡态的化学类似物结合来间接地分离催化剂。在另一个具体实例中,可以通过选择与底物的共价键形成(例如,使用与亲和标签连接的底物)或通过切割(例如通过选择破坏特定键,从而从固体支持物释放文库的催化成员的能力)来进行直接分离。在另一个具体实例中,选择的酶促反应可以关联次级信号反应,其包括但不限于颜色变化、荧光或发光。
分离新催化剂的这种方法具有优于催化抗体技术的至少两个重要优点(在schultz等人,j.chem.engng.new.86(1990))中综述)。首先,在催化抗体技术中,初始库通常限于免疫球蛋白折叠;相比之下,dna-rna-蛋白质融合物的起始文库可以是完全随机的,或者可以包括但不限于已知的酶促结构或蛋白质支架的变体。此外,催化抗体的分离通常依赖于结合过渡态反应类似物的初步选择,随后对活性抗体进行费力筛选;再次,相比之下,如先前使用rna文库证明的,使用dna-rna-蛋白质融合文库方法可以直接选择催化作用。在分离蛋白质酶的替代方法中,可以组合过渡态-类似物和直接选择方法。
通过该方法获得的酶是非常有价值的。例如,目前迫切需要新型和有效的工业催化剂,其允许开发改进的化学方法。本公开的主要优点是选择可以在任意条件下进行,并不限于例如体内条件。因此,本公开有助于分离新的酶或现有酶的改进变体,其可以进行高度特异性的转化(从而最小化不需要的副产物的形成),同时在预定的环境(例如升高的温度、压力或溶剂浓度的环境)中发挥功能。
5.16在微芯片形式中使用rna-蛋白质融合物
“dna芯片”由固定化寡核苷酸或cdna或基因组dna的克隆片段的空间定义阵列组成,并具有诸如快速测序和转录谱分析等应用。通过将dna-rna融合物(例如从细胞dna或rna库产生)的混合物退火到这样的dna芯片,可以产生“rna展示芯片”,其中对应于一个固定化序列的每个点(spot)能够退火到其在dna-rna融合库中相应的dna序列。通过这种方法,相应的rna由于其与其自身dna的连接而以空间定义的方式被固定化,并且含有dna序列组的芯片展示相应的rna组。
rna的这种有序展示具有许多用途。例如,它们代表了用于鉴定rna与其它分子之间的以前未知的相互作用的强大工具。可以使用固定在任何合适的固体支持物上的核酸(包括rna,但优选dna)的阵列进行rna展示技术。示例性的固体支持物可以由诸如玻璃(例如玻璃板),硅或硅玻璃(例如微芯片)或金(例如金板)等材料制成。将核酸连接到这种固体表面上的精确区域的方法,例如光刻方法,是本领域公知的,并且可用于产生固体支持物(例如dna芯片)。
本文所述的各种要素的所有组合都在本发明的范围内,除非本文另有说明或者另外与上下文明显矛盾。
本发明在以下部分中进行说明,这些部分列出以帮助理解本发明,并且不应被解释为以任何方式限制所附权利要求书中定义的本发明范围。
6.实施例
6.1
ctp-peg-dna合成:
专用试剂:
5-氨基烯丙基胞苷-5'-三磷酸(trilinkbiotechnologies)
5'硫醇修饰的dna寡聚物(integrateddnatechnologies)
马来酰亚胺,nhs异双官能peg接头(nanocs)
需要的缓冲液:
tlc缓冲液:95%乙醚5%丙酮
1x缀合缓冲液(pbs)
1.25x缀合缓冲液(pbs)
缓冲液a:20mmtris-hcl,ph8.5
缓冲液b:1mnacl,20mmtris-hcl,ph8.5
注意:为获得最佳结果,请确保制备dna-sh并准备好在步骤5中与ntp-peg-nh2结合。
寡核苷酸还原:
1.将dna寡聚物重新悬浮于pbsph7.0,约1nmole/μl。
2.混合tcep浆液,并取出相当于2x寡聚物体积的体积。将tcep浆液离心,除去上清液。然后用pbsph7.0洗涤tcep浆液。
3.然后将dna寡聚物加入到tcep浆液中,并在室温下持续混合孵育2小时。
4.然后将tcep浆液/dna寡聚物混合物转移到pallnanosep100微离心柱中,并以约10000rpm离心5分钟。滤液含有还原硫醇修饰的dna寡聚物。
缀合:
1.准备合适的tlc板,具有用于交联剂,ntp,t=0,t=30min,t=60min的泳道。
2.将10μl100mmaa-ctp溶液和40μl4.6875mm马来酰亚胺-peg3400-nhs在1.25xpbs,ph7.2溶液中合并。
3.将反应混合物在室温下孵育1小时或在4℃下孵育2小时。使用tlc监测反应。
4.使用g10sephadex微凝胶过滤柱除去多余的ctp。
5.将5×摩尔比的还原的dna-sh和脱盐的ntp-peg-与过量的dna-sh合并并混合。
6.在室温下将反应混合物孵育30分钟或在4℃下孵育2小时。用tlc监测反应。注意:一般来说,让反应进行数小时或过夜没有危害,尽管反应通常在指定时间内完成。为了在完成前停止缀合反应,加入含有还原半胱氨酸的缓冲液,其浓度比蛋白质-sh的巯基高几倍。
7.使用离子交换色谱法纯化。
离子交换缓冲液:
缓冲液a:20mmtris-hcl,ph8.5
缓冲液b:1mnacl,20mmtris-hcl,ph8.5
梯度0-100%,经30分钟
254nm检测ctp和260nm检测dna
然后可以使用质谱和page电泳表征各级分。
ctp-peg-dna至dna模板的链延伸:
invitrogenpfxpcr反应混合物
10x缓冲液5µl
ddh2o26.1µl
50mmmgso41µl
ntp-peg-dna引物16µm15µl
生物素化模板ssdna50µm1µl
dntp混合物10mm1.5µl
pfxdna聚合酶0.4µl
总计50µl
热循环仪程序
dna展示:
将生物素化的dsdna-peg-ctp模板与链霉抗生物素蛋白珠孵育5分钟并洗涤3次。
然后将珠重悬浮于:
100mmteaph7.627µl
50mmmgcl25µl
10mg/mlbsa5µl
100mmatp5µl
1单位胞苷酸激酶8µl
并在室温下孵育30分钟。
然后将珠洗涤3次,并重悬于
ddh2o39µl
10xt4连接酶缓冲液5µl
100mmatp5µl
1单位核苷5'-二磷酸激酶1µl
并在室温下孵育30分钟。
然后将珠在ddh2o中洗涤3次,分为无转录(-t)和转录(+t)等分试样,分别为1/3至2/3比例(1-t反应,1+t反应和1+trna酶反应)。
然后将+t珠重悬浮于epicentret7flash转录试剂盒反应混合物中:
depc处理的ddh206.3µl
10x转录缓冲液2µl
100mmatp1.8µl
100mmgtp1.8µl
100mmctp1.8µl
100mmutp1.8µl
100mmdtt2µl
rna酶抑制剂0.5µl
t7rna聚合酶2µl
总体积20µl
并在37℃下孵育30分钟。
然后将+t等分试样分为2份(1+t反应和1+t和rna酶反应)。将样品-t、+t和+trna酶重悬于19μlrna酶缓冲液(1mmmgcl2,0.5mmcacl2,10mmtrisph7.5)。向-t和+t加入1μlddh2o,向+trna酶样品加入1μl10μg/mlrna酶a。然后将所有样品在37℃下孵育15分钟,然后在ddh2o中洗涤3次。然后通过在3μl95%甲酰胺中在90℃下处理2分钟,从珠洗脱样品-t、+t和+trna酶。然后将样品加载到7m脲10%page凝胶上进行分析(图4)。
本文提及的所有出版物以其整体并入本申请。在本文使用的术语和通过引用并入本文的出版物中使用的术语之间存在明显的冲突的情况下,应理解为本说明书提供准确定义。
尽管为了清楚和理解的目的,已经对前述发明进行了一些细节的描述,但本领域技术人员通过阅读本公开将了解,可以在不脱离所附权利要求书中的本发明的真实范围的情况下进行形式和细节上的各种改变。
参考文献
1.nucleicacidligandsus5696249a
2.yeastcellsurfacedisplayofproteinsandusesthereofus6699658b1
3.expressionofproteinsonbacterialsurfaceus5348867a
4.selectionofproteinsusingrna-proteinfusionsus6261804b1
5.ribosomecomplexesasselectionparticlesforinvitrodisplayandevolutionofproteinsus6620587b1
6.phagedisplayus8685893b2
7.expressionofproteinsonbacterialsurfaceus5348867a
8.invitropeptideexpressionlibraryus8679781b2
9.oligonucleotide-linkedmagneticparticlesandusesthereofus5512439a
10.benatuil,l.等(2010)."animprovedyeasttransformationmethodforthegenerationofverylargehumanantibodylibraries."proteinengineeringdesignandselection23(4):155-159.
11.coomber,d.等(2004).invitropeptideexpressionlibraray.
12.earhart,c.f.等(1993).expressionofproteinsonbacterialsurface.
13.he,m.和m.j.taussig(1998).ribosomecomplexesasselectionparticlesforinvitrodisplayandevolutionofproteins.
14.liu,r.等(2000).selectionofproteinsusingrna-proteinfusions.
15.osborne,s.e.和a.d.ellington(1997)."nucleicacidselectionandthechallengeofcombinatorialchemistry."chemicalreviews97(2):349-370.
16.scott,j.k.和g.p.smith(1990)."searchingforpeptideligandswithanepitopelibrary."science249(4967):386-390.
17.sidhu,s.s.等(2012).phagedisplay.
18.vanantwerp,j.j.和k.d.wittrup(2000)."fineaffinitydiscriminationbyyeastsurfacedisplayandflowcytometry."biotechnologyprogress16(1):31-37.
19.wang,j.等(2014)."particledisplay:aquantitativescreeningmethodforgeneratinghigh‐affinityaptamers."angewandtechemieinternationaledition53(19):4796-4801.
20.wilson,d.s.等(2001)."theuseofmrnadisplaytoselecthigh-affinityprotein-bindingpeptides."proceedingsofthenationalacademyofsciences98(7):3750-3755.
21.wittrup,k.d.等(2004).yeastcellsurfacedisplayofproteinsandusesthereof。
- 还没有人留言评论。精彩留言会获得点赞!