水稻内源双向表达启动子的分离克隆及功能分析的制作方法
本发明属于水稻基因工程技术领域。具体涉及水稻内源双向表达启动子的分离克隆及功能分析。
背景技术:
启动子是启动基因转录的一段DNA序列,作为转录水平上的重要调控元件,它能够有效调控下游基因在特定时期、空间和信号刺激下的转录过程。随着作物遗传改良的进程和功能基因组研究的发展,越来越多的作物性状受到关注,同时越来越多可用于性状改良的候选基因得到鉴定,候选基因的高效应用必须依靠它们在特定部位、生育期或者信号刺激下的高效表达。因此,启动子的多样性在基因工程中受到迫切的需求。
迄今为止,越来越多的单向启动子被克隆和鉴定,根据它们表达模式可分为组成型启动子、组织特异型启动子和诱导型启动子(McElroy D,Zhang W,Cao J,Wu R.Isolation of an efficient actin promoter for use in rice transformation.The Plant Cell,1990,2:163-171;Cai M,Wei J,Li X,Xu C,Wang S.A rice promoter containing both novel positive and negative cis-elements for regulation of green tissue-specific gene expression in transgenic plants.Plant Biotechnol J,2007,5:664-674;Vijayan J,Devanna BN,Singh NK,Sharma TR.Cloning and functional validation of early inducible Magnaporthe oryzae responsive CYP76M7promoter from rice.Front Plant Sci,2015,6:371)。双向启动子泛指相邻且转录方向相反呈“头对头结构”的基因对之间的序列,在基因工程中表现出更好的应用潜力(Trinklein ND et al.An abundance of bidirectional promoters in the human genome.Genome Res,2004,14:62-66;Yang S,Sleight SC,Sauro HM.Rationally designed bidirectional promoter improves the evolutionary stability of synthetic genetic circuits.Nucleic Acids Res,2013,41:e33-e33)。这一优势主要得益于双向启动子可以同时驱动两个靶基因表达,在载体构建和基因聚合过程中更加方便省时(Kumar S et al.A combinatorial bidirectional and bicistronic approach for coordinated multi-gene expression in corn.Plant Mol Biol,2015,87:341-353)。而且,在导入多个外源基因的同时,往往需要使它们维持相同或相似的表达模式以达到对特定性状进行改良的目的(Ha SH et al.Application of two bicistronic systems involving 2A and IRES sequences to the biosynthesis of carotenoids in rice endosperm.Plant Biotechnol J,2010,8:928-938;Ogo Y,Ozawa K,Ishimaru T,Murayama T,Takaiwa F.Transgenic rice seed synthesizing diverse flavonoids at high levels:a new platform for flavonoid production with associated health benefits.Plant Biotechnol J,2013,11:734-746)。然而,具有相同或相似表达模式的单向启动子数量有限,重复使用可能造成生物体内基因表达的沉默,同时也在载体构建过程中造成时间和成本的更大消耗(Peremarti A et al.Promoter diversity in multigene transformation.Plant Mol Biol,2010,73:363-378)。由于很多双向基因对存在共表达的现象,双向启动子5’和3’的表达模式在很多报道中是相似的(Huang Y,Xiao B,Xiong L.Characterization of a stress responsive proteinase inhibitor gene with positive effect in improving drought resistance in rice.Planta,2007,226:73-85;Chen W,Meaux JD,Lercher MJ.Co-expression of neighbouring genes in Arabidopsis:separating chromatin effects from direct interactions.BMC Genomics,2010,11:178;Didych D et al.Human PSENEN and U2AF1L4genes are concertedly regulated by a genuine bidirectional promoter.Gene,2013,515:34-41)。因此,双向启动子还能够克服相同表达模式单向启动子的缺乏问题。
由于启动子并不像基因编码区有产物可以限制其在进化过程中的变异,因此,除了保守的顺式调控元件之外,启动子的序列存在高度变异(Müller F,Demény MA,Tora L.New problems in RNA polymerase II transcription initiation:matching the diversity of core promoters with a variety of promoter recognition factors.J Biol Chem,2007,282:14685-14689)。所以,研究启动子的保守性可以通过其调控的下游基因入手。早在2002年,Adachi等人就发现人类基因组中“头对头结构”的基因对普遍存在(Adachi N,Lieber MR.Bidirectional gene organization:a common architectural feature of the human genome.Cell,2002,109:807-809)。其后,Li等人通过系统,发现“头对头结构”的基因对在不同物种中存在较高的保守性(Li Y et al.Systematic analysis of head-to-head gene organization:evolutionary conservation and potential biological relevance.PLoS Comput Biol,2006,2:e74)。2012年,Xu等人通过“头对头结构”的基因对在不同物种间的结构保守性来研究双向启动子的保守问题(Xu C,Chen J,Shen B.The preservation of bidirectional promoter architecture in eukaryotes:what is the driving force?BMC Syst Biol,2012,6:S21)。
根据已有报道,双向启动子的鉴定方法可分为两种,一种是利用生物信息学手段的鉴定方法,与之相对的则是利用实验手段的鉴定方法。生物信息学鉴定方法不尽相同,本质上都是通过开发不同的语言模型预测启动子中存在的转录调控区域、MOTIF的结合位点或者核小体缺失区域,筛选符合条件的双向启动子进行表达相关性和功能相关性分析(Wang Q et al.Searching for bidirectional promoters in Arabidopsis thaliana.BMC Bioinformatics,2009,10:S29;Claeys M,Storms V,Sun H,Michoel T,Marchal K.MotifSuite:workflow for probabilistic motif detection and assessment.Bioinformatics,2012,28:1931-1932;Zhang W et al.High-resolution mapping of open chromatin in the rice genome.Genome Res,2012,22:151-162)。而实验鉴定方法则落脚于检测启动子驱动报告基因的表达模式这一常规的启动子分析方法上面。一些研究是通过启动子正反驱动同一个报告基因进行鉴定(Huang Y,Xiao B,Xiong L.Characterization of a stress responsive proteinase inhibitor gene with positive effect in improving drought resistance in rice.Planta,2007,226:73-85), 而一些研究是通过启动子同时驱动两个报告基因进行鉴定(Léjard V,Rebours E,Meersseman C,Rocha D.Construction and validation of a novel dual reporter vector for studying mammalian bidirectional promoters.Plasmid,2014,74:1-8),也有结合以上两种方法同时鉴定的报道(Mishra RC,Grover A.Intergenic sequence between Arabidopsis caseinolytic protease B-cytoplasmic/heat shock protein100and choline kinase genes functions as a heat-inducible bidirectional promoter.Plant Physiol,2014,166:1646-1658)。通过实验手段克隆的双向启动子,绝大多数都依附于其调控基因的研究。迄今为止,鲜有将生物信息学手段和实验手段相结合,根据双向启动子的结构特点和表达模式进行规模化筛选和鉴定的报道。
水稻是我国主要的粮食作物。因此,水稻遗传改良的重要性和必要性不言而喻。多基因的导入是其中很关键的一步。前文已详细介绍双向启动子在多基因导入中的优势,所以水稻双向启动子的挖掘对于水稻遗传改良是必不可少的。同时,水稻中完备的基因组信息(Goff SA et al.A draft sequence of the rice genome(Oryza sativa L.ssp.japonica).Science,2002,296:92-100;Yu J et al.A draft sequence of the rice genome(Oryza sativa L.ssp.indica).Science,2002,296:79-92;Pan Y et al.Comparative BAC-based physical mapping of Oryza sativa ssp.indica var.93-11and evaluation of the two rice reference sequence assemblies.Plant J,2014,77:795-805)及较为清楚的基因表达信息(Wang L et al.A dynamic gene expression atlas covering the entire life cycle of rice.Plant J,2010,61:752-766)也为双向启动子的筛选和鉴定提供了极大的便利。
技术实现要素:
本发明的目的在于在于克服现有技术存在的缺陷,建立一个双向启动子的筛选、克隆及功能分析的完整方法,并为基因工程和分子育种提供高效的双向启动子资源。本发明结合MSU和RAP数据库中的RNA-seq数据以及CREP数据库中的表达谱芯片数据在水稻基因组中选取了候选双向启动子BIP1,并利用它同时驱动两个报告基因绿色荧光蛋白基因(以下简称GFP基因)和β-葡糖醛酸酶基因(以下简称GUS基因)在水稻中表达。对转基因水稻的GUS和GFP分析结果显示,BIP1启动子具有双向表达活性。然后我们对其进行5’和3’缺失分析,鉴定出其双向表达调控区段以及控制其5’和3’方向基本表达活性的区段。随后,利用生物信息学手段分析了BIP1在禾本科植物中的保守性。本发明为水稻双向启动子的分离克隆及功能分析提供了一个成功的方法。
本发明的技术方案如下所述:
首先以MSU和RAP数据库中的RNA-seq数据以及CREP数据库中的表达谱芯片数据为基础,根据双向启动子调控基因的位置信息和表达信息,选取了一个候选双向启动子BIP1,其核苷酸序列如序列表SEQ ID NO:1所示。通过设计特异引物,从水稻明恢63的基因组中扩增BIP1,并构建表达载体BIP1-DX2181。通过对转基因水稻的GUS和GFP分析试验,我们发现启动子BIP1在两个方向均呈现出组成型高效表达。然后对其进行5’和3’缺失分析,鉴定出其双向表达调控区段R1以及控制其3’方向基本表达活性的区段R2和5’方向基本表达活性的区段R3。随后,利用生物信息学手段分析了BIP1在禾本 科植物中的保守性。
本发明公开了双向启动子的筛选及分离克隆方法、转化载体的构建过程、水稻遗传转化过程和转化植物的GUS组织化学染色、GUS蛋白活性检测、GFP组织学分析、GFP定量分析和双向启动子的保守性分析等方法。
本发明的具体步骤是:
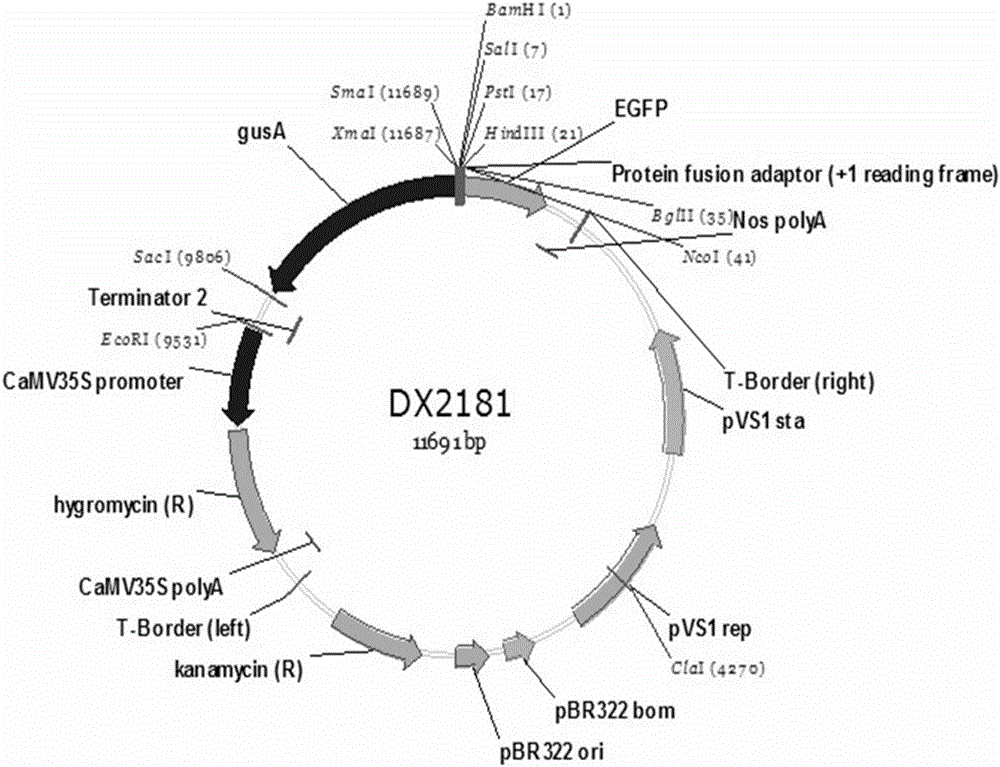
首先以MSU和RAP数据库中的RNA-seq数据以及CREP数据库中的表达谱芯片数据为基础,根据双向启动子调控基因的位置信息和表达信息,选取了一个候选双向启动子BIP1,其核苷酸序列如序列表SEQ ID NO:1所示。利用PCR法,以明恢63基因组DNA为模板,通过特异引物(表1)扩增BIP1。PCR扩增得到的BIP1经过TA克隆(购自Promega公司)后利用SP6和T7引物测序。经过测序验证的克隆通过Pst I单酶切将BIP1导入载体DX2181(载体构建图及多克隆位点等信息见图2),得到重组的表达载体BIP1-DX2181(见图3)。将重组的表达载体BIP1-DX2181通过电转化法导入农杆菌菌株EHA105。
将水稻品种中花11(来源于中国农业科学院作物科学研究所)的成熟种子消毒后诱导胚性愈伤组织。将含有表达载体BIP1-DX2181的农杆菌菌株EHA105与胚性愈伤组织进行共培养后,在含有50mg/L潮霉素的筛选培养基上进行抗性愈伤组织的筛选,经过两次筛选之后,挑取抗性愈伤转入分化培养基上进行分化,当分化的小苗长到2-3cm时,切掉原生根,放到生根培养基上进行生根,当新根生长到2cm左右时,炼苗后移栽到温室。这些再生小苗即为T0代转基因苗。
获得转基因植株后,通过GUS组织化学染色、GUS蛋白活性检测、GFP组织学分析及GFP定量分析,考察BIP1在水稻中的表达模式和表达强度(见图4、5)。检测结果表明:启动子BIP1在水稻中呈现出双向表达模式,且两个方向均为组成型高效表达。
设计特异引物(表1),用PCR法获得在BIP1的5’端和3’端缺失的不同长度的启动子(图6),并分别构建到载体DX2181(来源:澳大利亚CAMBIA实验室)上。以同样的方法转化水稻品种中花11。获得转基因植株后,通过GUS组织化学染色、GUS蛋白活性检测、GFP组织学分析及GFP定量分析(图7、8),鉴定出其双向表达调控区段R1以及控制其3’方向基本表达活性的区段R2和5’方向基本表达活性的区段R3。
随后,利用生物信息学手段分析发现:BIP1在六个禾本科植物(水稻、高粱、小米、二穗短柄草、玉米和小麦)中的水稻、高粱、二穗短柄草和玉米中均呈现出保守性。
本发明的优点在于:
(1)本发明结合生物信息学方法和实验手段,根据双向启动子调控基因的位置信息和表达信息,建立了一个双向启动子的筛选、克隆及功能分析的完整方法。
(2)本发明获得了一个在水稻中组成型高强度表达的双向启动子BIP1,为基因工程和分子育种提供了高效的双向启动子资源。
附图说明
序列表SEQ ID NO:1是本发明中双向启动子BIP1的核苷酸序列;序列长度为2237bp。
序列表SEQ ID NO:2是本发明中BIP1截短后的启动子BIP1-1F2R的核苷酸序列;序列长度为1240bp。 序列表SEQ ID NO:3是本发明中BIP1截短后的启动子BIP1-1F3R的核苷酸序列;序列长度为1099bp。序列表SEQ ID NO:4是本发明中BIP1截短后的启动子BIP1-2F1R的核苷酸序列;序列长度为1540bp。序列表SEQ ID NO:5是本发明中BIP1截短后的启动子BIP1-2F2R的核苷酸序列;序列长度为543bp。
图1:本发明的总体技术路线图。
图2:是DX2181载体结构示意图,本发明利用其骨架构建转化载体。
图3:是本发明构建的转化载体BIP1-DX2181结构示意图。该载体是在DX2181的基础上改造而来,将启动子BIP1插入质粒DX2181的多克隆位点构建而成。
图4:组织学分析BIP1驱动GUS基因和GFP基因在水稻不同组织中的表达情况。
图5:由BIP1驱动GUS基因和GFP基因的转化植株的不同组织中GUS蛋白活性检测和GFP定量分析结果。GFP relative expression level代表GFP的相对表达量;GUS activity(pMol 4-MU/min/mg protein)代表GUS酶活。
图6:是本发明对BIP1进行5’和3’缺失分析的示意图。
图7:组织学分析不同BIP1缺失启动子驱动GUS基因和GFP基因在水稻不同组织中的表达情况。
图8:由不同BIP1缺失启动子驱动GUS基因和GFP基因分别在转化植株各组织或器官中GUS蛋白活性检测和GFP定量分析结果。附图标记说明:图8中A图显示叶片的结果;图8中B图显示的是叶鞘的结果;图8中C图显示的是穗的结果;图8中D图显示的是茎杆的结果;图8中E图显示的是种子中的结果;图8中F图显示的是根的结果。GFP relative expression level代表GFP的相对表达量;GUS activity(pMol4-MU/min/mg protein)代表GUS酶活。
具体实施方式
实施例1:植物表达载体的构建
如无特别说明,本说明书中所列参考方法及相应分子生物学常规操作,参考:【J.萨姆布鲁克等,《分子克隆实验指南(第二版)》,中译本,科学出版社,北京,1996版】的相关信息。
本发明首先以MSU和RAP数据库中的RNA-seq数据以及CREP数据库中的表达谱芯片数据来选取候选的双向启动子。选取原则基于:1、符合相邻且转录方向相反呈“头对头结构”的基因对;2、RNA-seq数据中,基因对的最大表达数值同时高于10;表达谱芯片数据中,基因对的最大表达数值同时高于5000;3、基因对在95个样品的RNA-seq数据中的表达相关性高于0.4。依据以上原则选取了一组基因对,并分离它们的基因间区,即为候选双向启动子。将其命名为BIP1,其核苷酸序列如序列表SEQ ID NO:1所示。利用PCR法,以明恢63基因组DNA为模板,通过特异引物(表1)扩增BIP1。PCR扩增得到的BIP1经过TA克隆(Promega公司)后利用SP6和T7引物测序。经过测序验证的克隆通过Pst I单酶切将BIP1导入载体DX2181(载体图及多克隆位点等信息见图2),得到重组的表达载体BIP1-DX2181(见图3)。将表达载体BIP1-DX2181通过电转化法导入农杆菌菌株EHA105,将转化后的农杆菌菌株EHA105菌株于-70℃下保存待用。
表1本发明中使用的引物序列
表1的说明:a下划线序列表示酶切位点。
实施例2:农杆菌介导的遗传转化
农杆菌介导的遗传转化方法主要参照华中农业大学作物遗传改良国家重点实验室发表的“农杆菌介导的遗传转化操作手册”所示的方法(林拥军等,2002)。转化受体为水稻品种中花11(为常规品种,来源于中国农业科学院作物科学研究所)的成熟种子所诱导产生的胚性愈伤组织。经过预培养、侵染、共培养、筛选得到具有潮霉素抗性的愈伤,再经过分化、生根、练苗和移栽,得到转基因植株。本发明的遗传转化的主要步骤、培养基及其配制的方法如下所述:
(1)农杆菌介导的遗传转化步骤
1)愈伤诱导
a.将成熟的中花11水稻种子去壳,然后依次用70%的乙醇处理1分钟,0.15%氯化汞(HgCl2)种子表面消毒15分钟;
b.用灭菌水洗种子4-5次;
c.将种子放在诱导培养基上;
d.将接种后的培养基置于黑暗处培养4周,温度25±1℃。
2)愈伤继代
挑选亮黄色、紧实且相对干燥的胚性愈伤,放于继代培养基上黑暗下培养2周,温度25±1℃。
3)预培养
挑选紧实且相对干燥的胚性愈伤,放于预培养基上黑暗下培养2周,温度25±1℃。
4)农杆菌培养
a.在带有对应抗性选择的LA培养基上预培养农杆菌EHA105(该菌株来自CAMBIA公司商业化的农杆菌菌株)两天,温度28℃;
b.将农杆菌转移至悬浮培养基里,28℃摇床上培养2-3小时。
5)农杆菌侵染
a.将预培养的愈伤转移至灭好菌的瓶子内;
b.调节农杆菌的悬浮液至OD600 0.8-1.0;
c.将愈伤在农杆菌悬浮液中浸泡30分钟;
d.转移愈伤至灭菌好的滤纸上吸干;然后放在共培养基上培养3天,温度19-20℃。
6)愈伤洗涤和选择培养
a.灭菌水洗涤愈伤至看不见农杆菌;
b.浸泡在含400mg/L羧苄青霉素(CN)的灭菌水中30分钟;
c.转移愈伤至灭菌好的滤纸上吸干;
d.转移愈伤至选择培养基上选择培养2-3次,每次2周。
7)分化
将生长旺盛的抗性愈伤转入分化培养基中,放置于光照培养室中进行光照培养至分化出再生小苗。
8)生根
待再生小苗的芽长至2-3cm高时即可进行生根。用剪刀和镊子将再生植株在分化培养基上长出的根清除干净,将再生植株芽的下部插入生根培养基中,放置于光照培养室培养直至长出白色的新根。
9)炼苗及移栽
当新根生长到2cm左右时,可进行炼苗:将生根培养基的封口膜揭掉,加入适量自来水,在光照培养室继续培养3天。移栽:洗掉根上的残留培养基,将具有良好根系的幼苗转入温室,同时在最初的几天保持水分湿润。
(2)主要溶液配方
1)N6培养基大量元素母液(按照10倍浓缩液(10X)配制):
将上述试剂逐一溶解,然后室温下用蒸馏水定容至1000ml。
2)N6培养基微量元素母液(按照100倍浓缩液(100X)配制:
将上述试剂在室温下溶解并用蒸馏水定容至1000ml。
3)铁盐(Fe2-EDTA)贮存液(按照100X浓缩液配制):
将3.73g乙二胺四乙酸二钠(Na2EDTA·2H2O)和2.78g FeSO4·7H2O分别溶解,混合并用蒸馏水定容至1000ml,至70℃温浴2小时,4℃保存备用。
4)维生素贮存液(按照100X浓缩液配制):
加蒸馏水定容至1000ml,4℃保存备用。
5)MS培养基大量元素母液(按照10X浓缩液配制):
将上述试剂在室温下溶解,并用蒸馏水定容至1000ml。
6)MS培养基微量元素母液(按照100X浓缩液配制):
将上述试剂在室温下溶解,并用蒸馏水定容至1000ml。
7)2,4-D贮存液(1mg/ml)的配制:
称取2,4-D 100mg,用1ml 1N氢氧化钾溶解5分钟,然后加10ml蒸馏水溶解完全后定容至100ml,于室温下保存。
8)6-BA贮存液(1mg/ml)的配制:
称取6-BA 100mg,用1ml 1N氢氧化钾溶解5分钟,然后加10ml蒸馏水溶解完全后定容至100ml,室温保存。
9)萘乙酸(NAA)贮存液(1mg/ml)的配制:
称取NAA 100mg,用1ml 1N氢氧化钾溶解5分钟,然后加10ml蒸馏水溶解完全后定容至100ml,4℃保存备用。
10)吲哚乙酸(IAA)贮存液(1mg/ml)的配制:
称取IAA 100mg,用1ml 1N氢氧化钾溶解5分钟,然后加10ml蒸馏水溶解完全后定容至100ml,4℃保存备用。
11)葡萄糖贮存液(0.5g/ml)的配制:
称取葡萄糖125g,然后用蒸馏水溶解定容至250ml,灭菌后4℃保存备用。
12)AS贮存液的配制:
称取AS 0.392g,加入DMSO 10ml溶解,分装至1.5ml离心管内,4℃保存备用。
13)1N氢氧化钾贮存液配制:
称取氢氧化钾5.6g,用蒸馏水溶解定容至100ml,室温保存备用。
(3)用于水稻遗传转化的培养基配方
1)诱导培养基
加蒸馏水至900ml,1N氢氧化钾调节pH值到5.9,煮沸并定容至1000ml,分装到50ml三角瓶(25ml/瓶),封口后按常规方法灭菌(例如121℃下灭菌25分钟,下述的培养基灭菌方法与本培养基的灭菌方法相同)。
2)继代培养基
加蒸馏水至900ml,1N氢氧化钾调节pH值到5.9,煮沸并定容至1000ml,分装到50ml三角瓶(25ml/瓶),封口,按上述方法灭菌。
3)预培养基
加蒸馏水至250ml,1N氢氧化钾调节pH值到5.6,封口,按上述方法灭菌。
使用前加热溶解培养基并加入5ml葡萄糖贮存液和250ul AS贮存液,分装倒入培养皿中(25ml/皿)。
4)共培养基
加蒸馏水至250ml,1N氢氧化钾调节pH值到5.6,封口,按上述方法灭菌。
使用前加热溶解培养基并加入5ml葡萄糖贮存液和250ul AS贮存液,分装倒入培养皿中(25ml/每皿)。
5)悬浮培养基
加蒸馏水至100ml,调节pH值到5.4,分装到两个100ml的三角瓶中,封口,按上述方法灭菌。
使用前加入1ml无菌葡萄糖贮存液和100ul AS贮存液。
6)选择培养基
加蒸馏水至250ml,调节pH值到6.0,封口,按上述方法灭菌。
使用前溶解培养基,加入250ul HN(50mg/ml)和400ul CN(250mg/ml)分装倒入培养皿中(25ml/皿)。(注:第一次选择培养基羧苄青霉素浓度为400mg/l,第二次及以后选择培养基羧苄青霉素浓度为250mg/l)。
7)预分化培养基
加蒸馏水至250ml,1N氢氧化钾调节pH值到5.9,封口,按上述方法灭菌。
使用前溶解培养基,250ul HN(50mg/ml)250ul CN(250mg/ml),分装倒入培养皿中(25ml/皿)。
8)分化培养基
加蒸馏水至900ml,1N氢氧化钾调节pH值到6.0。
煮沸并用蒸馏水定容至1000ml,分装到50ml三角瓶(50ml/瓶),封口,按上述方法灭菌。
9)生根培养基
加蒸馏水至900ml,用1N氢氧化钾调节pH值到5.8。
煮沸并用蒸馏水定容至1000ml,分装到生根管中(25ml/管),封口,按上述方法灭菌。
实施例3:利用PCR法检测转基因阳性植株
将转化苗移入温室后,待其返青,然后分单株取1-2cm幼嫩叶片,抽提其基因组DNA,以基因组DNA为模板,利用PCR法检测阳性植株。扩增片段为报告基因gus的部分片段,片段大小为699bp。引物序列为GUS-F:GGGCGAACAGTTCCTGATTA,GUS-R:AACGTATCCACGCCGTATTC。PCR反应条件:94℃ 5min,94℃50sec,57℃40sec,72℃50sec,30个循环,72℃7min。PCR产物经0.8%琼脂糖胶电泳检测。对所有T0代植株进行PCR检测,根据检测结果剔除假阳性的转化植株。
利用小量叶片基因组DNA的提取方法提取基因组DNA:取适量幼嫩叶片,加800μl 1.5×CTAB(1.5×CTAB配方:1.5%CTAB、75mM Tris-HCl、15mM EDTA及1.05M NaCl)研磨,转入1.5ml离心管中;65℃水浴30min;加入600μL氯仿/异戊醇(体积比为24:1)上下颠倒数次(约15min),下层液相呈深绿色为止;室温下12000r/min离心10min;取500μL上清于一新1.5ml离心管,加入预冷的95%乙醇1mL,混匀后置-20℃,30min;室温下12000r/min离心10min,去上清,用75%乙醇浸洗沉淀,自然干燥;加入100μL ddH2O溶解,备用。
实施例4:启动子BIP1在水稻各组织中的表达模式
取表达载体BIP1-DX2181阳性转化植株(参考实施例3)的不同组织(包括:根、叶片、叶鞘、茎杆、穗及种子)切成约0.5CM长度的适当大小,浸入约200μl的GUS染液,37℃过夜,然后用75%酒精脱色,观察是否有蓝色出现。染色液的配方参照Jefferson等报道的方法(Jefferson,R.A.,Kavanagh,T.A.and Bevan,M.W.GUS fusions:β-glucuronidase as a sensitive and versatile gene fusion marker in higher plants.EMBO J.1987,6,3901-3907)。
表达载体BIP1-DX2181阳性转化植株的GFP组织学分析通过荧光显微镜进行。取T0代阳性转化植株的各个组织(叶片、叶鞘、茎杆、根、穗及种子)在荧光显微镜(Leica MZ16F)的GFP2档下观察,并利用Leica Application Suite software拍照。
检测结果显示:本发明分离的启动子BIP1在水稻中呈现出双向表达模式,且两个方向均为组成型高效表达(见图4)。
实施例5:启动子BIP1在水稻不同组织中的表达强度分析
取表达载体BIP1-DX2181阳性转化植株的不同组织(包括:根、叶片、叶鞘、茎杆、穗及种子)并抽提总蛋白,然后测定其GUS活性。总蛋白的抽提及浓度测定参考Bradford的方法(Bradford,M.M.A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding.Anal.Biochem.1976,72,248-254),GUS活性测定参考Xu的方法(Xu,L.et al.Isolation of the endosperm-specific LPAAT gene promoter from coconut(Cocos nucifera L.)and its functional analysis in transgenic rice plants.Plant Cell Rep.2010,29,1061-1068)。
BIP1-DX2181阳性转化植株不同组织的GFP表达量利用quantitative real-time PCR(qRT-PCR)进行检测。转基因植株不同组织的总RNA抽提方法和具体步骤如下(采用北京全式金生物科技有限公司的TransZol,具体操作步骤参考其说明书)。进行质量分析和浓度检测后,使用invitrogen公司的SSIII试剂盒进行反转录合成用于real-time PCR的cDNA第一链。qRT-PCR使用宝生物工程大连有限公司的SYBR Green Master Mix试剂盒。所用引物序列为:目的基因:GFP-F:5’-ATCCGCCACAACATCGAGGA-3’;GFP-R:5’-TCGTCCATGCCGAGAGTGAT-3’;内参基因:GAPDH-F:5’-CTGCAACTCAGAAGACCGTTG-3’; GAPDH-R:5’-CCTGTTGTCACCCTGGAAGTC-3’。反应在ABI 7500PCR仪上进行。实验结果分析方法采用2-ΔΔCT法。
BIP1-DX2181转基因植株的GUS及GFP报告基因定量检测的结果(图5)显示,启动子BIP1的3’方向的表达活性在种子中最高,其GUS活性达到17806±2108pmol 4-MU/min/mg protein;其次是根,GUS活性为14769±1782pmol 4-MU/min/mg protein;接下来是茎杆,叶鞘,穗和叶片,GUS活性分别为9825±1510,8681±834,7380±895和6092±875pmol 4-MU/min/mg protein。启动子BIP1的5’方向的表达活性在穗中最高,是叶片中表达活性的3.4倍;而根,叶鞘,茎杆和种子中GFP的表达量分别是叶片中的2.8,2.5,1.4和1.1倍。
实施例6:启动子BIP1的5’和3’缺失分析
为了寻找双向启动子BIP1的双向表达调控区域,对它进行5’和3’缺失分析。以测序验证的TA-BIP1为模板,利用PCR法,设计特异引物(表1)来构建BIP1的一系列5’和3’缺失启动子(图6)。载体构建、水稻遗传转化及转基因植株的GUS和GFP相关分析方法均与上述实施例相同。不同BIP1缺失启动子驱动GUS基因和GFP基因在水稻不同组织中的表达情况见图7。不同BIP1缺失启动子驱动GUS基因和GFP基因的转化植株的不同组织中GUS蛋白活性检测和GFP定量分析结果见图8。
结果显示,BIP1-1F2R转基因植株各组织的GUS活性远低于BIP1转基因植株,尤其是根,茎杆,种子和穗的GUS活性低于BIP1转基因植株相应组织的10%。由此可以推断,Region 1能够大幅度提升BIP1的3’方向的转录活性。与此同时,BIP1-1F2R转基因植株各组织的GFP表达量也明显低于BIP1转基因植株。以上结果表明,Region 1是一个双向转录增强区段。进一步截短BIP1的3’端导致BIP1-1F3R转基因植株的GUS活性完全丧失,而BIP1-1F3R转基因植株各组织的GFP表达量与BIP1-1F2R转基因植株相比并无明显降低。该结果表明,Region 2调控BIP1的3’方向的基本转录活性,而与5’方向的转录活性无关。BIP1-2F1R和BIP1-2F2R转基因植株均检测不到GFP表达,说明Region 3的缺失导致BIP1的5’方向的转录活性完全丧失。BIP1-2F1R转基因植株的GUS分析结果显示,Region 3的缺失对于BIP1在大多数组织中3’方向的表达活性影响微弱,但对启动子在根中3’方向的表达活性却影响很大,BIP1-2F1R转基因植株根的GUS活性明显低于BIP1转基因植株。上述结果表明,Region 3调控BIP1的5’方向的基本转录活性,同时,它还能够正向调控BIP1的3’方向在根中的转录活性。
实施例7:启动子BIP1在禾本科植物中的保守性分析
由于启动子并不像基因编码区有产物可以限制其在进化过程中的变异,因此,除了保守的顺式调控元件之外,启动子的序列存在高度变异。故研究启动子的保守性可以通过其调控的下游基因入手。本发明利用Ensembl Plants数据库(http://plants.ensembl.org/index.html)中的信息分析四组基因对在禾本科植物中“头对头结构”的保守性。若一组基因对在另一物种中均存在同源基因,且同源基因的排列方式仍然呈现“头对头结构”,则将调控它们的启动子视为保守的双向启动子(c-BIP);反之,若同源基因的排列方式不呈现“头对头结构”,则将调控它们的启动子视为非保守的双向启动子(n-BIP)。
表2启动子BIP1在不同禾本科植物间的保守性分析
表2的结果显示,本发明克隆的启动子BIP1在六个禾本科植物中的4个物种:水稻、高粱、二穗短柄草和玉米中均为保守,表明它调控的两个基因在进化过程中相对保守,在多数物种中均维持了这样的“头对头结构”。同时,受到双向启动子BIP1调控的两个基因,LOC_Os02g42314和LOC_Os02g42320的功能注释分别为泛素偶联酶和蛋白酶,都属于保守的结构基因。而且,它们在功能上也存在一定的相关性,进一步佐证其共进化的关系。
本发明获得了一个在水稻中组成型高强度表达的双向启动子BIP1,为基因工程和分子育种提供了高效的双向启动子资源。同时,本发明结合生物信息学方法和实验手段,根据双向启动子调控基因的位置信息和表达信息,建立了一个双向启动子的筛选、克隆及功能分析的完整方法。
- 还没有人留言评论。精彩留言会获得点赞!