一种醋酸甲羟孕酮分散片的有关物质及其分析检测方法与流程
本发明属于药物制剂分析领域,具体涉及一种醋酸甲羟孕酮分散片的有关物质及其分析检测方法。
背景技术:
醋酸甲羟孕酮分散片,适应症为用于不能手术,复发性或转移性激素依赖性肿瘤的姑息治疗或辅助治疗,如子宫内膜癌、肾癌、乳腺癌等。醋酸甲羟孕酮分散片的活性成分为醋酸甲羟孕酮,醋酸甲羟孕酮作为乳腺癌、子宫内膜癌等激素依赖性肿瘤的内分泌治疗药物,以及用于治疗月经不调、功能性子宫出血、子宫内膜异位症,亦可用于短效复方口服避孕片的孕激素成分,并可用于孕妇保胎。目前临床广泛应用并取得较满意的效果。同时,醋酸甲羟孕酮作为内分泌药物单用或与抗肿瘤药物合用治疗前列腺癌、肾癌、前列腺腺瘤也有提高疗效的作用。其他治疗乳腺癌、子宫内膜癌、前列腺癌、肾癌、前列腺腺瘤的化疗药物有一定的毒副作用,对病人机体产生伤害,但是醋酸甲羟孕酮的优点在于不但能够单独治疗或协同治疗,还能明显改善患者的厌食、体重减轻和恶病质等症状,从而改善患者的全身状况和生活质量。醋酸甲羟孕酮化学结构式如下:
技术实现要素:
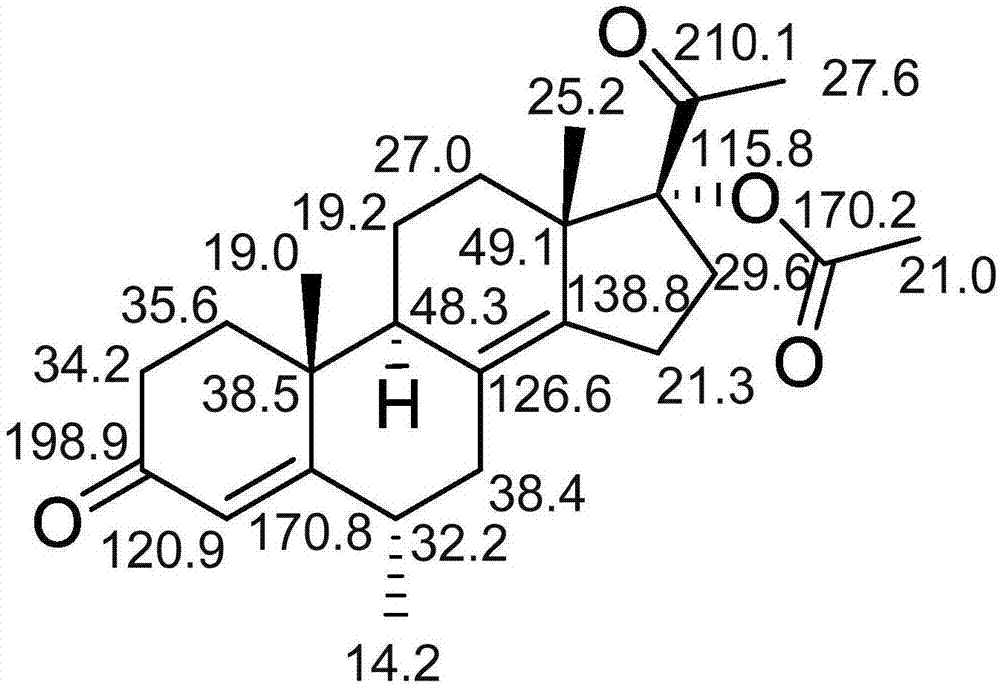
本发明首次提供了一种醋酸甲羟孕酮分散片的有关物质及其分析检测方法。本发明的目的之一是提供一种醋酸甲羟孕酮分散片中的有关物质,化学结构式如下:本发明的目的之二是提供上述有关物质的制备方法,包括如下步骤:步骤S1,高温破坏:将醋酸甲羟孕酮原料药置于100~120℃条件下破坏16~24小时;步骤S2,富集:将上述高温破坏样品溶于甲醇体积百分浓度为60%~70%的甲醇水溶液中,搅拌,抽滤,收集滤液,减压浓缩得浓缩物;步骤S3,粗品制备:将上述浓缩物用正相硅胶柱色谱分离,依次用体积比为30:1、15:1和8:1的二氯甲烷-甲醇混合溶剂梯度洗脱,收集8:1洗脱部分,浓缩得粗品;步骤S4,精制:将上述粗品用反相硅胶柱纯化,固定相为十八烷基硅烷键合硅胶,流动相为乙腈体积百分浓度为65%~75%的乙腈水溶液,等度洗脱,收集第8~12个柱体积洗脱液,浓缩,冷冻干燥即得所述有关物质。进一步地,所述的制备方法包括如下步骤:步骤S1,高温破坏:将醋酸甲羟孕酮原料药置于110℃条件下破坏20小时;步骤S2,富集:将上述高温破坏样品溶于甲醇体积百分浓度为65%的甲醇水溶液中,搅拌,抽滤,收集滤液,减压浓缩得浓缩物;步骤S3,粗品制备:将上述浓缩物用正相硅胶柱色谱分离,依次用体积比为30:1、15:1和8:1的二氯甲烷-甲醇混合溶剂梯度洗脱,收集8:1洗脱部分,浓缩得粗品;步骤S4,精制:将上述粗品用反相硅胶柱纯化,固定相为十八烷基硅烷键合硅胶,流动相为乙腈体积百分浓度为70%的乙腈水溶液,等度洗脱,收集第9~11个柱体积洗脱液,浓缩,冷冻干燥即得所述有关物质。本发明的目的之三是提供上述有关物质的分析方法,包括如下参数:色谱柱:XtimateC18(4.6mm×250mm,5μm);流动相:A为乙腈,B为含0.03mol/L磷酸二氢钠的水溶液(磷酸调节pH值至2.8);梯度洗脱程序:0~6min,A30%;6~26min,A30%→80%;26~32min,A80%;32~35min,A80%→30%;35~40min,A30%;流动相流速:1.0mL·min-1;检测波长:240nm;柱温:35℃;进样量:10μL。本发明的目的之四是提供上述有关物质在醋酸甲羟孕酮分散片有关物质检查中作为对照品的用途。本发明的优点:该有关物质为首次报道,为发明人在醋酸甲羟孕酮分散片有关物质研究中意外发现并经结构确证,其在醋酸甲羟孕酮原料药和片剂中均可检测到,且含量大于万分之五,有必要对其进行分析控制。该有关物质与醋酸甲羟孕酮的色谱保留行为非常接近,传统等度色谱条件很难分析发现该有关物质的存在,因此,本发明还提供了该有关物质的分析检测方法。本发明有助于醋酸甲羟孕酮分散片的质量研究控制的进一步研究,如长期稳定性考察和毒理考察,以提高醋酸甲羟孕酮分散片的质量标准。附图说明图1为本发明有关物质氢谱数据归属图;图2为本发明有关物质碳谱数据归属图。具体实施方式下面结合实施例进一步说明本发明的实质性内容,但并不以此限定本发明保护范围。尽管参照较佳实施例对本发明作了详细说明,本领域的普通技术人员应当理解,可以对本发明的技术方案进行修改或者等同替换,而不脱离本发明技术方案的实质和范围。实施例1:有关物质的制备和结构确证取10g醋酸甲羟孕酮原料均匀摊在培养皿中,置于烘箱中,110℃破坏20小时。然后,将高温破坏样品溶于甲醇体积百分浓度为65%的甲醇-水溶液中,搅拌20分钟,用抽滤漏斗抽滤,收集滤液,减压浓缩得浓缩物6.2g;然后将浓缩物用10ml甲醇溶解,用15g粒度为100~200目的正相硅胶拌样,挥干溶剂,用150g粒度为200~300目的正相硅胶作为分离硅胶进行柱色谱分离,依次用体积比为30:1、15:1和8:1的二氯甲烷-甲醇混合溶剂梯度洗脱,每个比例洗脱10个柱体积,收集8:1洗脱部分,减压浓缩得粗品4.1g;然后将粗品用乙腈体积百分浓度为70%的乙腈水溶液溶解,用反相硅胶柱纯化,固定相为十八烷基硅烷键合硅胶,流动相为乙腈体积百分浓度为70%的乙腈-水,等度洗脱,收集第9~11个柱体积的洗脱液,减压浓缩,冷冻干燥得粉末2.1g。HPLC归一化纯度大于98%。结构确证:HR-ESIMS显示[M+H]+为m/z385.2408,结合核磁特征可得分子式为C24H32O4。氢谱数据及归属见图1(ppm,methanol-d4,500MHz);碳谱数据及归属见图2(ppm,methanol-d4,150MHz)。综合质谱和核磁数据,结合醋酸甲羟孕酮的结构确证数据可确证该物质的化学结构式如下所示:实施例2:有关物质的制备取10g醋酸甲羟孕酮原料均匀摊在培养皿中,置于烘箱中,100℃破坏24小时。然后,将高温破坏样品溶于甲醇体积百分浓度为60%的甲醇-水溶液中,搅拌20分钟,用抽滤漏斗抽滤,收集滤液,减压浓缩得浓缩物6.0g;然后将浓缩物用10ml甲醇溶解,用15g粒度为100~200目的正相硅胶拌样,挥干溶剂,用150g粒度为200~300目的正相硅胶作为分离硅胶进行柱色谱分离,依次用体积比为30:1、15:1和8:1的二氯甲烷-甲醇混合溶剂梯度洗脱,每个比例洗脱10个柱体积,收集8:1洗脱部分,减压浓缩得粗品4.0g;然后将粗品用乙腈体积百分浓度为65%的乙腈水溶液溶解,用反相硅胶柱纯化,固定相为十八烷基硅烷键合硅胶,流动相为乙腈体积百分浓度为65%的乙腈-水,等度洗脱,收集第10~12个柱体积的洗脱液,减压浓缩,冷冻干燥得粉末1.9g。HPLC归一化纯度大于98%。实施例3:有关物质的制备取10g醋酸甲羟孕酮原料均匀摊在培养皿中,置于烘箱中,120℃破坏16小时。然后,将高温破坏样品溶于甲醇体积百分浓度为70%的甲醇-水溶液中,搅拌20分钟,用抽滤漏斗抽滤,收集滤液,减压浓缩得浓缩物6.3g;然后将浓缩物用10ml甲醇溶解,用15g粒度为100~200目的正相硅胶拌样,挥干溶剂,用150g粒度为200~300目的正相硅胶作为分离硅胶进行柱色谱分离,依次用体积比为30:1、15:1和8:1的二氯甲烷-甲醇混合溶剂梯度洗脱,每个比例洗脱10个柱体积,收集8:1洗脱部分,减压浓缩得粗品4.2g;然后将粗品用乙腈体积百分浓度为75%的乙腈水溶液溶解,用反相硅胶柱纯化,固定相为十八烷基硅烷键合硅胶,流动相为乙腈体积百分浓度为75%的乙腈-水,等度洗脱,收集第8~10个柱体积的洗脱液,减压浓缩,冷冻干燥得粉末2.2g。HPLC归一化纯度大于98%。实施例4:醋酸甲羟孕酮分散片加速试验(40℃条件下30天)样品检测分析醋酸甲羟孕酮分散片:收集市售不同品牌的醋酸甲羟孕酮分散片(距生产日期3个月内)。对照品溶液制备:精密称取2mg本发明有关物质于200ml容量瓶中,用乙腈溶解定容,作为有关物质储备液;精密量取有关物质储备液1ml于20ml容量瓶中,用液相分析方法中初始比例的流动相稀释定容(约0.5μg/mL)。供试品溶液制备:取40℃条件下放置30天的醋酸甲羟孕酮分散片研磨成细粉,精密称取细粉适量(约含醋酸甲羟孕酮12.5mg)置于25ml容量瓶中,添加液相分析方法中初始比例的流动相,超声使醋酸甲羟孕酮溶解,放冷,稀释至刻度。对照溶液:精密量取供试品溶液0.5ml于200ml容量瓶中,用液相分析方法中初始比例的流动相稀释至刻度。液相色谱条件:高效液相色谱仪:Agilent1260,二元泵,VWD;色谱柱:XtimateC18(4.6mm×250mm,5μm);流动相:A为乙腈,B为含0.03mol/L磷酸二氢钠的水溶液(磷酸调节pH值至2.8);梯度洗脱程序:0~6min,A30%;6~26min,A30%→80%;26~32min,A80%;32~35min,A80%→30%;35~40min,A30%;流动相流速:1.0mL·min-1;检测波长:240nm;柱温:35℃;进样量:10μL。分别精密量取有关物质对照品溶液、供试品溶液和对照溶液10μL注入液相色谱仪分析。结果在北京嘉德制药有限公司生产的醋酸甲羟孕酮分散片(批号:20150403)和浙江仙琚制药股份有限公司生产的醋酸甲羟孕酮分散片(批号:20150108)中均检测到本发明提供的有关物质(保留时间为醋酸甲羟孕酮保留时间1.25倍),该有关物质的色谱峰与醋酸甲羟孕酮色谱峰分离度大于2.0,且峰纯度检查合格。不同品牌醋酸甲羟孕酮分散片中该有关物质的含量为0.05~0.15%,有必要列入醋酸甲羟孕酮分散片的质量研究控制中进行进一步研究,如长期稳定性考察和毒理考察,以提高醋酸甲羟孕酮分散片的质量标准。上述实施例的作用在于说明本发明的实质性内容,但并不以此限定本发明的保护范围。本领域的普通技术人员应当理解,可以对本发明的技术方案进行修改或者等同替换,而不脱离本发明技术方案的实质和保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1