一种含P/N/Si/S/Ni协同催化阻燃的金属配合物及其制备方法与流程
本发明涉及一种膨胀型金属类阻燃剂,具体是一种含P/N/Si/S/Ni协同催化阻燃的金属配合物及其制备方法。
背景技术:
随着科学技术的迅速发展,高分子材料品种数量和使用范围日益增大,但它们有一个共性就是易燃或可燃,而阻燃剂是一种能阻止材料引燃或抑制火焰传播的有效物质。因此,阻燃剂的设计与制备已成为阻燃高分子材料领域的研究热点问题而备受关注。参见:Chen,L.;Wang,Y.Z.Polym.Adv.Technol.,2010,21,1-19。
在阻燃剂的开发应用上,卤系阻燃剂因其添加量少、阻燃效果好等优势仍是目前世界上用量最大的有机类阻燃剂,但由于其在阻燃燃烧过程中会释放许多烟和腐蚀性有毒气体,对人类的生命安全和环境造成极大危害而逐渐被近年来兴起来的无卤(低卤)膨胀型阻燃剂所代替。参见:Laoutid,F.;Bonnaud,L.;Alexandre M.;Lopez-Cuesta,J.M.;Dubois,P.Mat.Sci.Eng.R.,2009,63,100-119;Liang,S.;Neisius,N.M.;Gaan,S.Prog.Org.Coat.,2013,76,1642-1665。
膨胀型有机阻燃剂由于集酸源、碳源、气源于一体膨胀阻燃体系而发挥磷、氮等阻燃元素间的协同阻燃之功效而使其在阻燃过程中具有稳定性好、不宜消失、对聚合物基体性能影响小等优势;但为了显著提高阻燃效果,则需阻燃剂的使用量要非常大而造成阻燃基体的物理性能明显不佳之问题。鉴于此,科研人员引入硅、硫等阻燃元素到膨胀型阻燃体系中来进一步提升其阻燃效果,其中硅元素的引入可在阻燃燃烧过程中材料表面形成Si-O或Si-C键的无机保护层,硫元素的引入类似于磷元素可作为酸源在燃烧过程中形成酸性物质催化保护炭层的快速生成,这些均对阻断基体的进一步燃烧和避免有毒气体的大量生成而起到很好的协同阻燃之功效,能够进一步提高阻燃剂阻燃效果和降低其使用量。参见:Nguyen,T.M.D.;Chang,S.C.;Condon,B.;Uchimiya,M.;Fortier,C.Polym.Adv.Technol.,2012,23,1555-1563;Zhao,P.H.;Zhang,M.;Wu,D.H.Korean J.Chem.Eng.,2013,30,1678-1690;Li,X.H.;Chen,H.Y.;Wang,W.T.;Zhao,P.H.Polym.Degrad.Stabil.,2015,120,193-202;Zhao,P.H.;Liu,S.N.;Xiong,K.K.;Wang,W.T.Fiber Polym.,2016,17,569-575。
同时,近年来科研人员将一些金属氧化物和金属盐复配到膨胀型阻燃体系中表现出良好的阻燃效果,于是我们考虑将金属元素直接引入到膨胀型阻然体系分子中,发挥类似于有机金属配合物作为大量有机反应的催化剂之功能,使其在阻燃燃烧过程中催化加快成炭反应进行并快速生成保护炭层而起到很好的隔热隔氧作用,表现出高效的阻燃效果。
技术实现要素:
针对以上技术背景,本发明为了解决卤系阻燃剂的有烟有毒等非环保性能以及膨胀型有机阻燃剂的阻燃效率欠佳、使用量大等问题,而提供了一种中间体II及其制备方法,以及该中间体II的目标产物——含P/N/Si/S/Ni协同催化阻燃的金属配合物及其制备方法。
本发明是通过以下技术方案实现的:一种含P/N/Si/S/Ni协同催化阻燃的金属配合物的中间体II,其结构式为:
该结构式中X为Cl,Br,I(碘)。
另外,本发明提供了一种金属配合物的中间体II的制备方法,该金属配合物的中间体II如上所述,该制备方法包括如下步骤:
(1)将3-氨基丙基三乙氧基硅烷、二苯基氯化磷、三乙胺分别依次加入到二氯甲烷中,其溶液室温过夜反应后,将粗产物经过砂芯过滤、旋蒸浓缩、四氢呋喃洗涤、二氯甲烷重结晶、真空干燥,得到中间体I;
(2)将二卤化镍和中间体I依次加入第一溶剂中,其溶液在25℃-35℃下反应0.5h后停止反应,将粗产物经过旋蒸浓缩、无水乙醇和二氯甲烷依次洗涤、无水甲醇重结晶、真空干燥,得到中间体II。
本发明所述中间体II的制备方法中,所述步骤(1)中使用二氯甲烷重结晶的原因是中间体I在二氯甲烷的溶解性很好而易达到饱和溶液状态,其加入量为本领域技术人员容易控制实现的。所述步骤(2)中二卤化镍为六水合二氯化镍、无水二溴化镍、无水二碘化镍作为反应原料,其目的是所生成的中间体II分别包含了反应活性明显不同的三个卤素(I>Br>Cl),这是由于Cl、Br、I的原子半径依次增大,与金属核形成的Ni-X键依次减弱,在反应过程中双硫醇的硫负离子进攻Ni核中心时Cl、Br、I的离去性依次增强(即反应活性依次增大),故生成目标金属配合物的产量依次提高。
所述步骤(2)中无水乙醇和二氯甲烷依次洗涤目的是无水乙醇除去少量可能未反应的二卤化镍和二氯甲烷出去少量未反应的中间体I(过量),其加入量为本领域技术人员容易控制实现的。所述步骤(2)中使用无水甲醇重结晶的原因是中间体II在无水甲醇的溶解性最好而易达到饱和溶液状态,其加入量为本领域技术人员容易控制实现的。
具体实施时,为了提高中间体II的产率,优选的,所述3-氨基丙基三乙氧基硅烷、二苯基氯化磷、三乙胺的摩尔比为1:2.3:2.2;所述二卤化镍和中间体I的摩尔比为1:1.1。
进一步,所述第一溶剂为无水乙醇与二氯甲烷的混合物、无水甲醇与二氯甲烷的混合物。前述第一溶剂相对于其他溶剂,这两种混合溶剂相对于一般文献中所使用的单一反应溶剂来说,更能保证了各反应原料的充分溶解,让其反应过程在均相体系中进行,因为二卤化镍无机盐易溶于无水乙醇和无水甲醇中而难溶于一般有机溶剂,其它反应原料如3-氨基丙基三乙氧基硅烷和二苯基氯化磷在二氯甲烷中溶解性最好。
另外,所述二卤化镍为六水合二氯化镍、无水二溴化镍或无水二碘化镍。
本发明的另外一个目的是提供上述中间体II的目标产物——含P/N/Si/S/Ni协同催化阻燃的金属配合物,其结构式为:
本发明为了更清楚的说明其技术内容,提供了一种含P/N/Si/S/Ni协同催化阻燃的金属配合物的制备方法,该金属配合物如前述,该制备方法所采用的中间体II如前述,该制备方法包括如下步骤:
将中间体II溶于第二溶剂中,然后室温搅拌下依次滴加2-硫代-1,3-丙二硫醇和缚酸剂,反应0.5-1.5h后,将粗产物经过砂芯过滤、旋蒸浓缩、无水乙醇、四氢呋喃和乙醚依次洗涤、二氯甲烷重结晶、真空干燥,得到金属配合物。
本发明所述金属配合物的制备方法中,无水甲醇、四氢呋喃和乙醚依次洗涤目的是:无水乙醇除去少量可能未反应的中间体II,四氢呋喃除去少量剩余的缚酸剂盐酸盐,二氯甲烷出去少量未反应的中间体I(过量),各加入量为本领域技术人员容易控制实现的。使用二氯甲烷重结晶的原因是金属配合物在二氯甲烷的溶解性最好而易达到饱和溶液状态,其加入量为本领域技术人员容易控制实现的。
所述金属配合物的合成路线如下:
为了提高本发明金属配合物的产率,优选的,所述中间体II、2-硫代-1,3-丙二硫醇和缚酸剂的摩尔比为1:1.1~1.3:2~4。
优选的,所述的第二溶剂为二氯甲烷、四氢呋喃、二氯甲烷与乙醚的混合物、四氢呋喃与乙醚的混合物。前述第二溶剂相对于其他溶剂,这几种溶剂能充分保证各反应原料的充分溶解,让其反应过程在均相体系中进行,因为中间体II在二氯甲烷和四氢呋喃中的溶解性最大而其它有机溶剂难溶,原料双硫醇在乙醚中溶解性最好同时也能溶于二氯甲烷和四氢呋喃。
具体实施时,所述的缚酸剂为三乙胺、吡啶和二异丙基乙胺。
本发明相对于现有技术具有如下有益效果:
①本发明提供的中间体II可用于制备多种含P/N/Si/S/Ni协同催化阻燃的金属配合物;
②所述金属配合物同时将磷、氮、硅、硫四种阻燃元素集合到一个分子中并配位于金属原子,充分发挥多种阻燃元素协同膨胀阻燃性和金属元素催化成炭阻燃性,大大提高了其作为阻燃剂的阻燃效果;
③该阻燃剂的分子中无卤素原子并含有易反应分解的硅氧烷基,故其在燃烧过程中无有毒气体和无甲醛的释放,恰恰满足了现代阻燃剂阻燃燃烧过程的绿色环保之要求;同时其在使用量较少的情况下可用于对各种棉织物基体进行耐久性阻燃处理并能达到良好的预期阻燃效果;
④本发明的合成工艺条件温和而稳定、反应过程简单而易控制、原料廉价而易得、后处理方便而快捷,可制备多种[SiP2NS2M]类金属配合物以扩大膨胀型金属类阻燃剂在阻燃高分子材料领域的应用。
附图说明
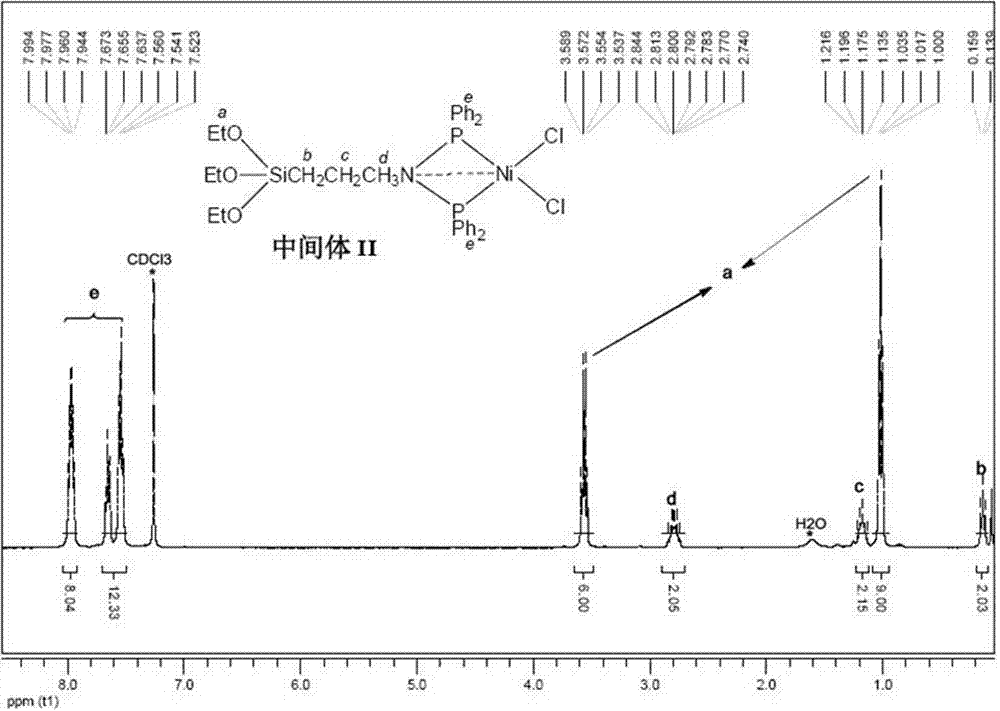
图1为本发明实施例1所述中间体II的核磁共振氢谱图。
图2为本发明实施例1所述中间体II的核磁共振磷谱图。
图3为本发明实施例1所述金属配合物的核磁共振氢谱图。
图4为本发明实施例1所述金属配合物的核磁共振磷谱图。
具体实施方式
实施例1
一种含P/N/Si/S/Ni协同催化阻燃的金属配合物的制备方法为:
(1)在30mL的二氯甲烷中,先加入2.3mL(10mmol)的3-氨基丙基三乙氧基硅烷,再缓慢加入4.1mL(23mmol)的二苯基氯化磷,随后缓慢加入3.1mL(22mmol)的三乙胺;室温搅拌过夜反应后,得到淡黄色悬浊液,将其通过砂芯过滤除去三乙胺盐酸盐固体得到淡黄色液体,将其旋蒸除去二氯甲烷得到固体粗产物;最后将其经过四氢呋喃洗涤、二氯甲烷重结晶和真空干燥得淡白色固体,即为中间体I(产率为89%);熔点为66-68℃。
(2)在35mL无水乙醇+10mL二氯甲烷的混合溶剂中,先加入1.19g(5.0mmol)的六水合二氯化镍剧烈搅拌至完全溶解,再加入3.25g(5.5mmol)的中间体I后,在25℃-35℃下反应0.5h后停止反应得到砖红色浑浊液,将其除去混合溶剂得到粘稠固体,最后将其经过无水乙醇和二氯甲烷依次洗涤、无水甲醇重结晶、真空干燥得砖红色粉末固体,即为中间体II(产率为90%);熔点大于250℃。
中间体II(X=Cl)的结构表征数据如下:1H-NMR(CD2Cl2,400MHz,TMS)δH/ppm:7.99-7.94(m,8H,4C6H5-o),7.67-7.52(m,12H,4C6H5-m,p),3.56(q,J=7.2Hz,6H,3OCH2CH3),2.81-2.74(m,2H,NCH2CH2CH2Si),1.22-1.14(m,2H,NCH2CH2CH2Si),1.02(t,J=7.2Hz,9H,3OCH2CH3),0.14(t,J=8.0Hz,2H,NCH2CH2CH2Si).31P-NMR(CDCl3,162MHz,85%H3PO4)δp/ppm:41.46(s).
(3)在氮气气氛保护下,将1.44g(2.0mmol)含双氯的中间体II(砖红色粉末)加入35mL二氯甲烷中并搅拌溶解,再缓慢注入0.2mL(2.4mmol)的2-硫代-1,3-丙二硫醇,随后缓慢滴加0.8mL(6.0mmol)的三乙胺缚酸剂,室温搅拌反应过程中红色反应液变为黑色溶液,室温反应1h后停止反应得黑色浑浊液,将其通过砂芯过滤除去三乙胺盐酸盐得到黑色液体,旋蒸除去二氯甲烷得粘稠固体;最后将其经过无水甲醇、四氢呋喃和乙醚依次洗涤、二氯甲烷重结晶、真空干燥得到黑色固体,即为金属配合物(产率为85%)。
金属配合物的结构表征数据如下:1H-NMR(CD2Cl2,400MHz,TMS)δH/ppm:8.00-7.81(m,8H,4C6H5-o),7.67-7.46(m,12H,4C6H5-m,p),3.56(q,J=7.2Hz,6H,3OCH2CH3),2.90-2.80(m,2H,NCH2CH2CH2Si),1.25-1.16(m,4H,2SCH2),1.03(t,J=7.2Hz,9H,3OCH2CH3),0.88-0.85(m,2H,NCH2CH2CH2Si),0.14(t,J=8.0Hz,2H,NCH2CH2CH2Si).31P-NMR(CDCl3,162MHz,85%H3PO4)δp/ppm:63.13(s).
实施例2
一种含P/N/Si/S/Ni协同催化阻燃的金属配合物的制备方法为:
(1)在30mL的二氯甲烷中,先加入2.3mL(10mmol)的3-氨基丙基三乙氧基硅烷,再缓慢加入4.1mL(23mmol)的二苯基氯化磷,随后缓慢加入3.1mL(22mmol)的三乙胺;室温搅拌过夜反应后,得到淡黄色悬浊液,将其通过砂芯过滤除去三乙胺盐酸盐固体得到淡黄色液体,将其旋蒸除去二氯甲烷得到固体粗产物;最后将其经过四氢呋喃洗涤、二氯甲烷重结晶和真空干燥得淡白色固体,即为中间体I(产率为88%)。
(2)在35mL无水乙醇+10mL二氯甲烷的混合溶剂中,先加入1.10g(5.0mmol)的无水二溴化镍剧烈搅拌至完全溶解,再加入3.25g(5.5mmol)的中间体I后,在25℃-35℃下反应0.5h后停止反应得到红色浑浊液,将其除去混合溶剂得到粘稠固体,最后将其经过无水乙醇和二氯甲烷依次洗涤、无水甲醇重结晶、真空干燥得红色粉末固体,即为中间体II(产率为87%)。
(3)在氮气气氛保护下,将1.62g(2.0mmol)含双溴的中间体II(红色粉末)加入35mL二氯甲烷中并搅拌溶解,再缓慢注入0.2mL(2.4mmol)的2-硫代-1,3-丙二硫醇,随后缓慢滴加0.8mL(6.0mmol)的三乙胺缚酸剂,室温搅拌反应过程中红色反应液变为黑色溶液,室温反应1h后停止反应得黑色浑浊液,将其通过砂芯过滤除去三乙胺盐酸盐得到黑色液体,旋蒸除去二氯甲烷得粘稠固体;最后将其经过无水甲醇、四氢呋喃和乙醚依次洗涤、二氯甲烷重结晶、真空干燥得到黑色固体,即为金属配合物(产率为89%)。
实施例3
一种含P/N/Si/S/Ni协同催化阻燃的金属配合物的制备方法为:
(1)在30mL的二氯甲烷中,先加入2.3mL(10mmol)的3-氨基丙基三乙氧基硅烷,再缓慢加入4.1mL(23mmol)的二苯基氯化磷,随后缓慢加入3.1mL(22mmol)的三乙胺;室温搅拌过夜反应后,得到淡黄色悬浊液,将其通过砂芯过滤除去三乙胺盐酸盐固体得到淡黄色液体,将其旋蒸除去二氯甲烷得到固体粗产物;最后将其经过四氢呋喃洗涤、二氯甲烷重结晶和真空干燥得淡白色固体,即为中间体I(产率为91%)。
(2)在35mL无水甲醇+10mL二氯甲烷的混合溶剂中,先加入1.34g(5.0mmol)的无水二碘化镍剧烈搅拌至完全溶解,再加入3.25g(5.5mmol)的中间体I后,在25℃-35℃下反应0.5h后停止反应得到深红色浑浊液,将其除去混合溶剂得到粘稠固体,最后将其经过无水乙醇和二氯甲烷依次洗涤、无水甲醇重结晶、真空干燥得深红色粉末固体,即为中间体II(产率为85%)。
(3)在氮气气氛保护下,将1.73g(2.0mmol)含双碘的中间体II(深红色粉末)加入35mL二氯甲烷中并搅拌溶解,再缓慢注入0.2mL(2.4mmol)的2-硫代-1,3-丙二硫醇,随后缓慢滴加0.8mL(6.0mmol)的三乙胺缚酸剂,室温搅拌反应过程中红色反应液变为黑色溶液,室温反应1h后停止反应得黑色浑浊液,将其通过砂芯过滤除去三乙胺盐酸盐得到黑色液体,旋蒸除去二氯甲烷得粘稠固体;最后将其经过无水甲醇、四氢呋喃和乙醚依次洗涤、二氯甲烷重结晶、真空干燥得到黑色固体,即为金属配合物(产率为92%)。
实施例4
一种含P/N/Si/S/Ni协同催化阻燃的金属配合物的制备方法为:
(1)在50mL的二氯甲烷中,先加入4.6mL(20mmol)的3-氨基丙基三乙氧基硅烷,再缓慢加入8.2mL(46mmol)的二苯基氯化磷,随后缓慢加入6.2mL(44mmol)的三乙胺;室温搅拌过夜反应后,得到淡黄色悬浊液,将其通过砂芯过滤除去三乙胺盐酸盐固体得到淡黄色液体,将其旋蒸除去二氯甲烷得到固体粗产物;最后将其经过四氢呋喃洗涤、二氯甲烷重结晶和真空干燥得淡白色固体,即为中间体I(产率为85%)。
(2)在35mL无水甲醇+10mL二氯甲烷的混合溶剂中,先加入1.79g(7.5mmol)的六水合二氯化镍剧烈搅拌至完全溶解,再加入4.89g(8.3mmol)的中间体I后,在25℃-35℃下反应0.5h后停止反应得到砖红色浑浊液,将其除去混合溶剂得到粘稠固体,最后将其经过无水乙醇和二氯甲烷依次洗涤、无水甲醇重结晶、真空干燥得砖红色粉末固体,即为中间体II(产率为88%)。
(3)在氮气气氛保护下,将1.44g(2.0mmol)含双氯的中间体II(砖红色粉末)加入35mL四氢呋喃中并搅拌溶解,再缓慢注入0.18mL(2.2mmol)的2-硫代-1,3-丙二硫醇,随后缓慢滴加0.53mL(4.0mmol)的三乙胺缚酸剂,室温搅拌反应过程中红色反应液变为黑色溶液,室温反应1h后停止反应得黑色浑浊液,将其通过砂芯过滤除去三乙胺盐酸盐得到黑色液体,旋蒸除去二氯甲烷得粘稠固体;最后将其经过无水甲醇、四氢呋喃和乙醚依次洗涤、二氯甲烷重结晶、真空干燥得到黑色固体,即为金属配合物(产率为80%)。
实施例5
一种含P/N/Si/S/Ni协同催化阻燃的金属配合物的制备方法为:
(1)在35mL的二氯甲烷中,先加入3.45mL(15mmol)的3-氨基丙基三乙氧基硅烷,再缓慢加入6.20mL(35mmol)的二苯基氯化磷,随后缓慢加入4.70mL(33mmol)的三乙胺;室温搅拌过夜反应后,得到淡黄色悬浊液,将其通过砂芯过滤除去三乙胺盐酸盐固体得到淡黄色液体,将其旋蒸除去二氯甲烷得到固体粗产物;最后将其经过四氢呋喃洗涤、二氯甲烷重结晶和真空干燥得淡白色固体,即为中间体I(产率为83%)。
(2)在45mL无水甲醇+10mL二氯甲烷的混合溶剂中,先加入1.32g(6.0mmol)的无水二溴化镍剧烈搅拌至完全溶解,再加入3.90g(6.6mmol)的中间体I后,在25℃-35℃下反应0.5h后停止反应得到红色浑浊液,将其除去混合溶剂得到粘稠固体,最后将其经过无水乙醇和二氯甲烷依次洗涤、无水甲醇重结晶、真空干燥得红色粉末固体,即为中间体II(产率为85%)。
(3)在氮气气氛保护下,将1.62g(2.0mmol)含双溴的中间体II(红色粉末)加入30mL四氢呋喃+5mL乙醚中并搅拌溶解,再缓慢注入0.18mL(2.2mmol)的2-硫代-1,3-丙二硫醇,随后缓慢滴加0.53mL(4.0mmol)的吡啶缚酸剂,室温搅拌反应过程中红色反应液变为黑色溶液,室温反应1h后停止反应得黑色浑浊液,将其通过砂芯过滤除去吡啶盐酸盐得到黑色液体,旋蒸除去二氯甲烷得粘稠固体;最后将其经过无水甲醇、四氢呋喃和乙醚依次洗涤、二氯甲烷重结晶、真空干燥得到黑色固体,即为金属配合物(产率为84%)。
实施例6
一种含P/N/Si/S/Ni协同催化阻燃的金属配合物的制备方法为:
(1)在60mL的二氯甲烷中,先加入6.9mL(30mmol)的3-氨基丙基三乙氧基硅烷,再缓慢加入12.3mL(69mmol)的二苯基氯化磷,随后缓慢加入6.3mL(66mmol)的三乙胺;室温搅拌过夜反应后,得到淡黄色悬浊液,将其通过砂芯过滤除去三乙胺盐酸盐固体得到淡黄色液体,将其旋蒸除去二氯甲烷得到固体粗产物;最后将其经过四氢呋喃洗涤、二氯甲烷重结晶和真空干燥得淡白色固体,即为中间体I(产率为82%)。
(2)在45mL无水甲醇+10mL二氯甲烷的混合溶剂中,先加入1.61g(6.0mmol)的无水二碘化镍剧烈搅拌至完全溶解,再加入3.90g(6.6mmol)的中间体I后,在25℃-35℃下反应0.5h后停止反应得到深红色浑浊液,将其除去混合溶剂得到粘稠固体,最后将其经过无水乙醇和二氯甲烷依次洗涤、无水甲醇重结晶、真空干燥得深红色粉末固体,即为中间体II(产率为83%)。
(3)在氮气气氛保护下,将1.73g(2.0mmol)含双碘的中间体II(深红色粉末)加入30mL二氯甲烷+10mL乙醚的混合溶剂中并搅拌溶解,再缓慢注入0.21mL(2.2mmol)的2-硫代-1,3-丙二硫醇,随后缓慢滴加1.06mL(8.0mmol)的二异丙基乙胺缚酸剂,室温搅拌反应过程中红色反应液变为黑色溶液,室温反应1h后停止反应得黑色浑浊液,将其通过砂芯过滤除去二异丙基乙胺盐酸盐得到黑色液体,旋蒸除去二氯甲烷得粘稠固体;最后将其经过无水甲醇、四氢呋喃和乙醚依次洗涤、二氯甲烷重结晶、真空干燥得到黑色固体,即为金属配合物(产率为85%)。
- 还没有人留言评论。精彩留言会获得点赞!