核酸分子的生成的制作方法
本申请为分案申请,其原申请的申请日为2008年2月1日,申请号为200880003836.x,名称为“核酸分子的生成”。本发明在广义上涉及在增强型固相多核苷酸扩增后生成单链核酸分子的方法。本发明还提供了以单链及双链核酸分子标记固体基质的方法。用于生成单链核酸分子及用于进行扩增反应的试剂盒也形成本发明的一部分。本发明还提供了用于生成任选地以报告分子标记的单链核酸分子的扩增体系,以及它们尤其作为标签、引物和探针的用途。
背景技术:
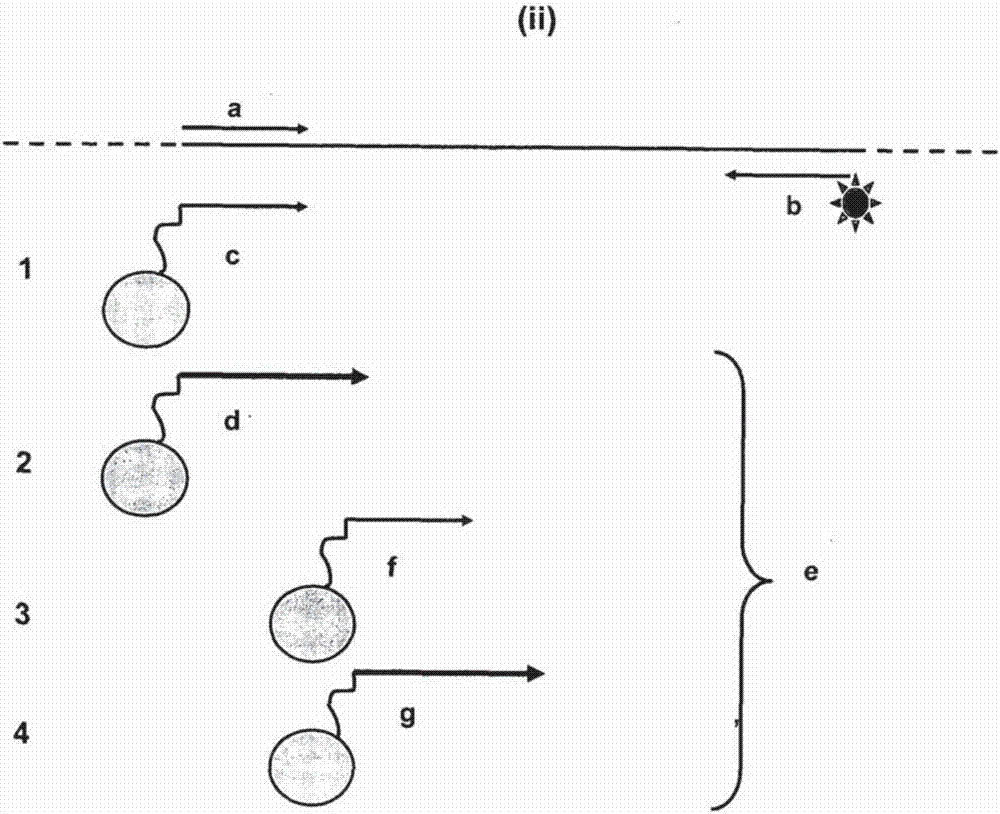
:在本说明书中参考的任何现有技术不是并且不能被认为是承认或以任何形式暗示,该现有技术在任何国家组成公知常识的一部分。通过将链分开或者通过去除双链体的一条链,可以将双链dna(dsdna)转换成单链dna(ssdna)。通过热方式或化学方式破坏链间键可以将双链体的链分开。去除一条链使得所需链被回收并消除其互补链,例如nikiforovetal.(美国专利no.5,518,900),其描述了通过以下方式修饰用于扩增的一个或多个引物:在待修饰引物的5’端整合硫代磷酸核苷酸衍生物,使得它抗外切核酸酶降解。在使用聚合酶链反应(pcr)扩增靶序列后,将dsdna进行外切核酸酶降解。未保护的链优选地被通过5’至3’的外切核酸酶消化,剩下包含另一条链的单链产物。类似策略使用了抗外切核酸酶的分枝引物(branchedprimer)(shchepinovetal,nuc.acids.res.25:4441-44541997)或λ外切核酸酶的携带5’磷酸的底物偏好性(higuchietal,nucl.acidsres.25:5685,1989)。非对称pcr(gyllenstenanderlich,proc.natl.acad.sci.usa85:7652-76561998;美国专利5,066,584)通过应用不平衡的引物对浓度使得一个引物处于限制浓度,在热循环过程中生成ssdna。这有利于由过量引物引发的ssdna产物。该方法具有的问题是持续合成能力(processivity)方面的固有限制,这是因为所使用的引物必须处于相对低的浓度。竞争性引物非对称pcr(gillespie,1997;美国专利申请08/628,417)采用在pcr热循环后及进一步热循环之前分开地添加竞争性引物,以生成ssdna。照这样,该方法需要繁琐的操作,这在特别是诊断环境下是不利的,因为污染风险、使用者的错误以及处理时间和成本增加。kaltenboecketal,biotechniques12:164-171,1992描述了一种通过以下方式产生ssdna的方法:最初进行pcr以生成dsdna,随后的分开反应是使用第一次pcr的产物作为模板并且采用一个引物进行第二次线性扩增。同样,该方法需要繁琐的操作。固相基质已经被以pcr产物标记,使用的是其中使一个引物缀合于固体表面上的对称pcr或非对称pcr,或者是通过其中使正向和反向引物直接缀合于固体表面上的“桥”pcr。这些方法中的每种由于动力学限制(有或无竞争性抑制效应的低效率底物浓度)均是相对低效率的。根据需要,缀合于固相上的dsdna产物可以被相对简单地通过化学或热变性转换为结合于固相上的ssdna产物。美国专利6,277,604描述了在非对称pcr中使用一个固定的引物和两个水相引物。至少一个所提供的水相引物的浓度是受限制的,以促进固定引物引发延伸事件。然而,这会导致低效率。明显需要开发更有效的方法,用于生成特定的单链核酸分子及用于标记固相载体。技术实现要素:在本说明书通篇及随后的权利要求书中,除非上下文中另外要求,否则词语“包含(comprise)”及变体如“包含(comprises)”或“包含(comprising)”应被理解为意指包括所述整数或者整数或步骤组,但不排除任何其他整数或整数组。本发明定义了一种用于生成单链(ss)多核苷酸,特别是特定ssdna分子并且增强固相多核苷酸扩增的方法。甚至更具体地,本发明采用了一种使用在特定退火条件下具有差异引发特性的引物的扩增反应。在存在允许的退火条件下进行一段时间的指数扩增之后,改变退火条件以有利于差异引发,有效地生成ssdna或包含其形式的核苷酸类似物。差异引物退火特性还可通过提供动力学优点的特定引物设计实现,或者通过允许的/非允许的退火及引物设计实现。因此,与水相引物相比,引发中固体支持引物的参与通过引物设计而得以提高。因此,以损害敏感性进行的非对称扩增对于在固相载体上获得高载量的扩增子不是必需的。本发明有利于无损失的且更敏感的固相扩增。扩增子相关信号在固相载体上的高加载被实现。本发明的方法可以在扩增反应器中进行,不需要另外的样品操作或处理。方便地(虽然不是必须地),至少一个引物被以能够提供识别信号的报告分子标记。本文的方法被称为增强型固相pcr(enhancedsolidphase-pcr)(“esp-pcr”)。因此,本发明考虑到了一种用于核酸的固相扩增的方法,其包括在实现固定引物延伸的反应条件下,将其上具有以连接子方式结合的引物的固相载体与包含至少一种核酸分子的样品接触,其中所述固定引物选自:(i)与水相引物有序列同一性但具有不同于固定引物的熔解温度的引物;和(ii)嵌套于两个水相引物之间的引物。在引物的上下文中提到“嵌套的”包括完全地或部分地嵌套的引物。所形成的固定核酸可以是ss或ds。然后,可以将所述固定ds核酸分子进行变性,以生成固定的ss核酸分子和水相ss核酸分子中的一种或两者。根据另一方面,本发明提供了一种在反应器中生成单链多核苷酸的方法,所述方法包括通过以下方式将双链的靶多核苷酸或其单链的衍生物进行指数扩增:在实现固定引物延伸的反应条件下,将其上具有以连接子方式结合的引物的固体基质与包含至少一种核酸分子的样品接触,其中所述固定引物选自:(i)与水相引物有序列同一性但具有不同于固定引物的熔解温度的引物;和(ii)嵌套于两个水相引物之间的引物;以有利于生成固定于所述固体基质的双链多核苷酸,然后使固定的多核苷酸处于变性条件以生成固定的单链多核苷酸及液相的单链多核苷酸。扩增反应可以是对称的、非对称的聚合酶链反应(pcr)、链替代扩增(sda)、连接酶链反应、切口限制性内切核酸酶(切口re),尤其是sda或全基因组扩增。方便地,至少一个水相引物被以能够提供识别信号的报告分子标记。上述在本文中被称为esp-pcr的方法是一种形式的分步扩增反应。这种分步的方案依赖于第一及第二退火条件或者引物设计,可以是“逐级上升的”或“逐级下降的”。差异退火条件的实例包括不同退火温度、引物和靶互补多核苷酸之间错配程度、无关或补足序列的存在,以及在靶核苷酸序列上不同互补位点内的引物设计。在一个实施方案中,一个或两个引物携带在同源性上与靶多核苷酸序列相关或不相关的“尾”核苷酸序列或“头”核苷酸序列,提供不同于另一个引物的熔解温度。因此,本发明还可以使用携带一种或多种无关或补足序列的引物进行,以协助降低核酸扩增偏差。本发明的方法具有许多应用,其中包括生成杂交反应的探针(例如rna印迹、dna印迹、克隆库筛选、原位杂交、荧光原位杂交(fish)和微阵列(如芯片、珠子或其他基于固体基质的分析))。本方法还可以与另一种改良的扩增方法结合使用,其中dsdna靶经5‘至3’外切核酸酶的消化以生成在本方法中使用的ssdna模板。本发明还考虑一种用于固相基质的直接多核苷酸标记的方法,例如在高通量测序方案或珠子微阵列应用之前的微阵列特征生成、用于基于非凝胶电泳的核酸分析的微滴定板标记、用于非基于凝胶电泳的遗传分析(如基于facs的遗传分析)的珠子标记以及乳液pcr中。包含反应器及试剂的试剂盒也是本发明的一部分。“标记”包括以报告分子例如化学发光分子或生物发光分子标记,以及/或者以核酸分子标记。因此,本发明还涉及这样的扩增体系,即其包含试剂组件、核酸组件、硬件组件和说明组件。该体系的组件相互之间相互作用,以生成单链多核苷酸产品。本发明还用于固相基质的直接多核苷酸标记,例如在高通量测序方案或珠子微阵列应用之前的微阵列或固体阵列特征生成、用于非基于凝胶电泳的核酸分析(如基于facs的遗传分析)的微滴定板标记以及乳液扩增中。本发明还可以被用于:在变性固定的ds核酸分子之后,生成固定于固相载体上的或者在水相中的ss核酸分子。因此,在一个具体的实施方案中,本发明考虑一种以单链多核苷酸标记固体基质的方法,所述方法包括使用在第一组退火条件下具有相似退火特性但在第二组退火条件下具有差异退火特性的正向及反向引物进行双链的靶多核苷酸或其单链的衍生物的指数扩增,将扩增的扩增产物与其上固定有至少一种这样的引物的固体基质或固体基质组合物接触,即该引物能够在第二组退火条件下与扩增产物的链退火,并且将退火条件转换成所述第二组条件,从而有利于基于单引物的扩增以生成固定于所述固体基质上的单链多核苷酸。在另一个实施方案中,本发明提供了一种以单链多核苷酸标记固体基质的方法,所述方法包括使用在第一组退火条件下具有相似退火特性但在第二组退火条件下具有差异退火特性的一对正向和反向引物对双链的靶多核苷酸或其单链的衍生物进行指数扩增,将扩增的扩增产物与其上固定有至少一种这样的引物的固体基质或固体基质组合物接触,即该引物能够在第二组退火条件下与扩增产物的链退火生成双链的多核苷酸,然后将双链的多核苷酸变性以生成固定于所述基质上的单链多核苷酸。还提供了一种用于标记固体基质的方法,其包括在实现固定引物延伸的反应条件下,将其上具有以连接子方式结合的引物的固相载体与包含至少一种核酸分子的样品接触,其中所述固定引物选自:(i)与水相引物有序列同一性但具有不同于固定引物的熔解温度的引物;和(ii)嵌套于两个水相引物之间的引物。提及“固体基质”包括可以固定核酸分子的固相、载体或者其他表面或珠子。上述方法包括这样的任选项,即将退火条件变成第一组条件,以使得“加入”单链多核苷酸以生成固定于固体基质上的双链多核苷酸。因此,本发明可扩展至一种生成用双链多核苷酸标记的固体基质或固体基质组合物的方法,所述方法包括使用在第一组退火条件下具有相似退火特性但在第二组退火条件下具有差异退火特性的一对正向和反向引物对双链的靶多核苷酸或其单链的衍生物进行指数扩增,将扩增的产物与其上固定有至少一种这样的引物的固体基质或固体基质组合物接触,即该引物能够在第二组退火条件下与扩增产物的链退火,并且将退火条件转换成第二组以生成固定于所述固体基质上的单链的多核苷酸;转换成第一组退火条件,借此在存在另一条引物的情况下,该单链的固定多核苷酸生成互补链,且所述固体基质包含固定的双链体多核苷酸。在又一实施方案中,所述双链体多核苷酸被通过例如化学热方式变性,以生成水相的单链多核苷酸或固定的单链多核苷酸。还提供了一种方法用于生成具有双链多核苷酸的固体基质或固体基质组合物,所述方法包括在实现固定引物延伸的反应条件下,通过将具有其上以连接子方式结合的引物的固相载体与包含至少一种核酸分子的样品接触,对双链的靶多核苷酸或其单链的衍生物进行指数扩增,其中所述固定引物选自:(i)与水相引物有序列同一性但具有不同于固定引物的熔解温度的引物;和(ii)嵌套于两个水相引物之间的引物。还可以使用嵌套的引物及允许的和非允许的条件。本说明书中使用的缩写在表1中被定义。表1缩写缩写定义dsdna双链的dnafacs荧光活化的细胞分拣fish荧光原位杂交pcr聚合酶链反应sda链替代扩增ssdna单链dna本文使用的序列标识符的总结显示于表2中。表2序列标识符序列标识符序列seqidno:1寡核苷酸(transprobe)seqidno:2cto3模板生成正向引物seqidno:3cto3模板生成反向引物seqidno:4ngo模板生成正向引物seqidno:5ngo模板生成反向引物seqidno:6ngps模板生成正向引物seqidno:7ngps模板生成反向引物seqidno:8cto3“水相”正向引物seqidno:9cto3“水相”反向引物seqidno:10cto3sp-pcr固相载体引物seqidno:11cto3esp-pcr固相载体引物seqidno:12ngo“水相”正向引物seqidno:13ngo“水相”反向引物seqidno:14ngosp-pcr固相载体引物seqidno:15ngoesp-pcr固相载体引物seqidno:16ngps“水相”正向引物seqidno:17ngps“水相”反向引物seqidno:18ngpssp-pcr固相载体引物seqidno:19ngpsesp-pcr固相载体引物seqidno:20淋病奈瑟菌opa引物seqidno:21淋病奈瑟菌opa反向引物seqidno:22沙眼衣原体隐蔽性质粒orf3引物seqidno:23沙眼衣原体反向引物附图说明某些附图包含彩色的表示或部分。彩色照片可以通过请求从专利所有人或者可以从合适的专利局获得。如果从专利局获得可能会收取费用。图1(i)和(ii)的图式表示为(i)增强型固相pcr(esp-pcr)方案,用于将单链的或双链的扩增子加载至具有(ii)固相载体引物设计(1至4)的固相载体上。参考实施例1的详细描述,(i):a–代表“正向”pcr引物;b–代表“反向”引物;c–使用例如正向和反向引物的指数扩增(如pcr);d–固相载体引物[见(ii)];e–许多ds拷贝/固相载体;f–许多ss拷贝/固相载体;g–(可选)具有不允许引物a和b的“分步的”退火条件的循环;h–(可选)“加入”;(ii):a–代表“正向”pcr引物;b–表“反向”引物;c–现有技术固相pcr固相载体引物设计;d–例如3-引发的延伸、较高tm;e–增强型固相pcr固相载体引物设计;f–例如嵌套的或部分嵌套的;g–例如嵌套的或部分嵌套的、较高tm。图2的图式表示为于使用标准(a)sp-pcr相比,使用(b)esp-pcr提高固相载体引发的机制。各反应混合物中所包括的“水相”正向(箭头)及反向引物的浓度不受限制,同时包含固相载体引物(连接于箭头上的球)。用“水相”引物进行常规pcr以生成扩增子(虚线)。在这些循环中固相载体引物也引发延伸反应,使得将产物加载至所述固相载体表面上。然而,在标准sp-pcr中,固相载体引物的参与受到匹配序列(a)的“水相”引物的竞争抑制,以产生相对弱的扩增子加载。(a)中的垂直线指示受抑制的延伸。esp-pcr(b)可通过应用具有相对高tm的嵌套固相载体引物避免这种抑制。虚线代表延伸事件。(a)在标准sp-pcr固相载体引物序列中的固相载体引发可匹配其对应的“水相”引物;(a)–固相载体引发被其“水相”引物对应物竞争掉;(b)–单链扩增子;(b)esp-pcr引物中的固相载体引发被嵌套,以通过利用不同的结合位点及“水相”引物结合及聚合酶结合之间的滞后,降低它和“水相”引物之间对于与扩增子结合的竞争。而且有较高的tm,以升高其有效浓度(a)–固相载体引发是竞争性的;(b)-单链的扩增子。具体实施方式本发明涉及一种改良的扩增反应,该反应有利于生成单链的(ss)多核苷酸,特别是固定于固相载体的ssdna,或者从固定的双链(ds)多核苷酸生成的固定形式或水相形式的ssdna。本发明被预测的部分用途是扩增反应中的引物,所述引物在一组退火条件(其中所述引物被认为是“平衡的”并且所述退火条件被认为是“互相允许的”)下具有相似的退火特征,并且在另一组退火条件(其中所述引物被认为是“不平衡的”并且所述退火条件被认为是“差异允许的”)下具有不同的或差异的或不相似的退火特征。此外或另外,不同的退火条件可源自引物设计,所述引物设计对固相载体引发赋予优于水相寡核苷酸引发的不同动力学优点。因此,本发明的一个方面考虑一种用于固相扩增的方法,其包括在实现固定引物延伸的反应条件下,将具有以连接子方式结合于固相载体的引物的固相载体与包含至少一种核酸分子的样品接触,其中所述固定引物选自:(i)与水相引物有序列同一性但具有不同于固定引物的熔解温度的引物;和(ii)嵌套于两个水相引物之间的引物。在引物的上下文中提到“嵌套的”包括完全地或部分地嵌套的引物。提及“共有序列同一性”包括基本上同一的,以及至少80%的序列同一性,例如至少80%、81%、82%、83%、84%、85%、86%、87%、88%、89%、90%、91%、92%、93%、94%、95%、96%、97%、98%、99%或100%的同一性。如上文指出的,正向引物和反向引物就退火特性而言可以被描述为“平衡的”或“非平衡的”,退火条件就对引物的效应而言可以被认为互相允许的或差异允许的。类似地,引物设计可有利于差异允许的引物。因此,平衡引物或非平衡引物是指,在给定的一组条件或给定的一组引物——即互相允许的退火条件或互相允许的引物下,两个引物均结合或者杂交以形成具有类似效率的双链体。而引物在差异允许的退火条件下(即在另一组条件下)是非平衡的,这是因为该条件可能有利于双链体形成及引发。因此,本发明可以被进一步限定为一种在反应器中生成固定于固相载体上的单链多核苷酸的方法,所述方法包括通过在互相允许的条件下使用一对平衡的正向引物和反向引物来在所述容器中进行固定于固相载体上的靶多核苷酸的扩增反应,然后将退火条件转变成差异允许的条件借此引物变为非平衡的,并且继续该反应以生成单链的多核苷酸产物,然后失活聚合酶活性以基本上阻止双链多核苷酸的形成。在另一个实施方案中,在固相载体上生成ds多核苷酸,然后使所述ds多核苷酸处于变性条件(例如通过化学途径或热途径)以生成固定的ss多核苷酸或水相多核苷酸。因此,本发明提供了一种以单链的多核苷酸标记固体基质的方法,所述方法包括使用在第一组退火条件下具有相似退火特性但在第二组退火条件下具有差异退火特性的正向及反向引物进行双链的靶多核苷酸或其单链的衍生物的指数扩增,将扩增的扩增产物与其上固定有至少一种这样的引物的固体基质或固体基质组合物接触,即该引物能够在第二组退火条件下与扩增产物的链退火生成双链的多核苷酸,然后将双链的多核苷酸变性以生成固定于所述固体基质上的单链多核苷酸。变性可通过包括化学途径或热途径的任何途径进行。另一方面提供了一种在反应器中生成单链多核苷酸的方法,所述方法包括通过以下方式将双链的靶多核苷酸或其单链的衍生物进行指数扩增:在实现固定引物延伸的反应条件下,将其上具有以连接子方式结合的引物的固体基质与包含至少一种核酸分子的样品接触,其中所述固定引物选自:(i)与水相引物有序列同一性但具有不同于固定引物的熔解温度的引物;和(ii)嵌套于两个水相引物之间的引物;以有利于生成固定于所述固体基质上的双链多核苷酸,然后使固定的多核苷酸处于变性条件下以生成固定的单链多核苷酸及液相的单链多核苷酸。然后可以进行另一步骤,该步骤是使所产生的混合物经受相分离途径处理。在另一个实施方案中,提供了一种在反应器中生成单链的多核苷酸的方法,所述方法包括在实现固定引物延伸的反应条件下,将其上具有以连接子方式结合的引物的固相载体与包含至少一种核酸分子的样品接触,其中所述固定引物选自:(i)与水相引物有序列同一性但具有不同于固定引物的熔解温度的引物;和(ii)嵌套于两个水相引物之间的引物。优选地,所述多核苷酸为dna,因此本发明具体地涉及ssdna的生成。然而,虽然起始材料优选地是dsdna,本发明还可以使用mrna(所述mrna被转换成dsdna或ssdna)或者使用ssdna。再者,可以在固相载体上生成dsdna,然后使其处于变性条件下以生成固定的ssdna或水相ssdna。本发明的另一个方面提供了一种在反应器中生成ssdna的方法,所述方法包括使用在第一组退火条件(互相允许条件)下具有相似的(平衡的)退火引物但在第二组退火条件(差异允许条件)下具有差异退火特性的一对正向和反向引物在反应器中进行从dsdna或mrna靶标到固定的靶ssdna模板的扩增反应,其中所述扩增被允许在互相允许的退火条件下进行;将条件转换成差异允许条件以使引物失去平衡,从而在基本上仅存在单引物的情况下促进线性扩增,以生成ssdna产物;并且使任何聚合酶活性失活以基本上降低或者阻止dsdna形成。本发明的又一方面为一种在反应器中生成ssdna的方法,所述方法包括在实现固定引物延伸的反应条件下,将其上具有以连接子方式结合的引物的固相载体与包含至少一种核酸分子的反应器中的样品接触,其中所述固定引物选自:(i)与水相引物有序列同一性但具有不同于固定引物的熔解温度的引物;和(ii)嵌套于两个水相引物之间的引物。差异允许的与互相允许的退火条件或引物与例如以下因素有关:错配程度、在引物上存在“头”或“尾”无关核苷酸序列、在靶模板上形成双链体的杂交位点或者存在其他使得能够逐级上升或逐级下降以生成非平衡的引物条件的特征。本发明的方法可以被认为是对扩增反应的改良以生成特定产物。所述改良为使用这样的引物,即所述引物依赖于退火条件的允许水平被差异地或互相地平衡。因此,本发明考虑一种改良的扩增反应,其中使用正向引物及反向引物指数地扩增从双链多核苷酸例如dsdna生成的或者直接地从mrna生成的或者经由cdna模板生成的单链多核苷酸例如ssdna,其中所述改良包括选择在第一组退火条件下具有相似退火特性且在第二组退火条件下具有差异退火特性的引物,使得将退火条件转变成第二组有助于在存在单引物的情况下的扩增,使得产生大量单链多核苷酸例如ssdna。此外或另外,引物设计被用于将固定引物嵌套于两个水相引物之间。本发明还提供了一种在反应器中生成单链多核苷酸的方法,所述方法包括通过以下方式将双链的靶多核苷酸或其单链的衍生物进行指数扩增:在实现固定引物延伸的反应条件下,将其上具有以连接子方式结合的引物的固体基质与包含至少一种核酸分子的样品接触,其中所述固定引物选自:(i)与水相引物有序列同一性但具有不同于固定引物的熔解温度的引物;和(ii)嵌套于两个水相引物之间的引物;以有利于生成固定于所述固体基质上的双链多核苷酸,然后使所述固定的多核苷酸处于变性条件以生成固定的单链多核苷酸及水相的单链多核苷酸。本发明的该方面通过整合一种缀合于3’模板结合区上的非引物结合区5’无关核苷酸序列,解决了潜在的扩增偏差的问题。这样的一个或两个引物整合了5’外来序列(尾序列),所述外来序列作为锁止(clamp)使扩增同系物间的扩增效率趋于平均。所述尾序列在同源性上可以与靶序列相关或不相关。又一方面还提供了一种在反应器中生成单链多核苷酸的方法,包括在实现固定引物延伸的反应条件下,将其上具有以连接子方式结合的引物的固相载体与包含至少一种核酸分子的样品接触,其中所述固定引物选自:(i)与水相引物有序列同一性但具有不同于固定引物的熔解温度的引物;和(ii)嵌套于两个水相引物之间的引物。对本文的方法的变化包括使用嵌套的引物及差异熔解条件。本发明具有多项应用,包括生成用于杂交反应(例如rna印迹、dna印迹、克隆文库筛查、原位杂交、荧光原位杂交(fish)及微阵列分析例如使用芯片、珠子或其他固体基质)的单链核苷酸探针。所述引物还可以被能够提供可识别信号的报告分子标记。例如,可以将荧光剂、发磷光剂、化学发光剂或放射性标记物整合至引物中。另外的标记物包括但不限于生物素-dutp、藻红蛋白-dutp、荧光素-dutp和[α-32p]-dutp,包括它们所有可能的异构体。还可以应用基于酶的检测法或化学检测法。本发明的方法还可以与其他扩增改良结合使用。例如应用外切核酸酶特别是5’至3’外切核酸酶以从dsdna靶生成ssdna模板的扩增体系。因此,本发明的另一方面提供了一种在反应器中生成ssdna的方法,所述方法包括:获得其中包含凹的5’端或平端的待扩增的dsdna靶标或dsdna靶标的区域,将所述dsdna和5’至3’外切核酸酶与dna等温扩增所需试剂一块孵育,其中所述5’至3’外切核酸酶产生包含dsdna每条链的3’至5’ssdna片段的ssdna模板,所述ssdna模板被用作扩增的模板;使用在第一组退火条件(互相允许条件)下具有相似的(平衡的)退火引物但在第二组退火条件(差异允许条件)下具有差异退火特性的一对正向和反向引物在所述ssdna上进行扩增反应,其中所述扩增被允许在互相允许的退火条件下进行;将所述条件转换成差异允许条件以使引物失去平衡,从而在基本上仅存在单引物的情况下促进线性扩增以生成ssdna产物;并且使任何聚合酶活性失活以基本上降低或者阻止dsdna形成。本发明的另一方面提供了一种在反应器中生成ssdna的方法,所述方法包括:获得其中包含凹的5’端或平端的待扩增的dsdna靶标或dsdna靶标的区域,将所述dsdna和5’至3’外切核酸酶与dna等温扩增所需试剂一块孵育,其中所述5’至3’外切核酸酶产生包含dsdna每条链的3’至5’ssdna片段的ssdna模板,所述ssdna模板被用作扩增的模板;并且通过以下方式在所述ssdna上进行扩增反应:在实现固定引物延伸的反应条件下,将其上具有以连接子方式结合的引物的固相载体与包含至少一种核酸分子的样品接触,其中所述固定引物选自:(i)与水相引物有序列同一性但具有不同于固定引物的熔解温度的引物;和(ii)嵌套于两个水相引物之间的引物。又再者,本文描述的方法可以与降低扩增偏差的方法结合使用。因此,另一方面提供了一种在反应器中生成单链多核苷酸的方法,所述方法包括:使用在第一组退火条件下具有相似退火特性但在第二组退火条件下具有差异退火特性的一对正向和反向引物在反应器中进行靶多核苷酸的扩增反应,其中所述扩增被允许在所述第一组退火条件下进行;将退火条件转换成第二组退火条件,以在基本上仅存在所述引物对中的一个引物的情况下促进线性应用,然后使扩增反应中的聚合酶活性失活以基本上阻止双链多核苷酸形成,其中所述扩增步骤包括使用正向引物及反向引物将所述多核苷酸靶标的核酸模板进行扩增,其中至少一个引物包含缀合于3’模板结合引物区上的5’无关核苷酸序列,其中所述无关核苷酸序列在引发起始后被整合至扩增产物中。该步骤可降低扩增差异。如上文指出的,所述5’外来序列与所述靶序列可以相关或不相关。本发明还用于固相基质的直接多核苷酸标记,例如在高通量测序方案或珠子微阵列应用之前的微阵列或固体阵列特征生成、用于基于非凝胶电泳的核酸分析的微滴定板标记、用于基于非凝胶电泳的核酸分析(如基于facs的遗传分析)的珠子标记以及乳液扩增中。因此,在一个具体实施方案中,本发明考虑一种以单链多核苷酸标记固体基质的方法,所述方法包括:使用在第一组退火条件下具有相似退火特性但在第二组退火条件下具有差异退火特性的正向及反向引物进行双链的靶多核苷酸或其单链的衍生物的指数扩增;将扩增的扩增子产物与其上具有至少一种这样的引物的固体基质或固体基质组合物接触,即该引物能够在第二组退火条件下与扩增子产物的链退火;并且将退火条件转换成所述第二组条件,从而有利于基于单引物的扩增以生成固定于所述固体基质上的单链多核苷酸。上述方法还可任选地包括将退火条件转换成第一组条件,以使得“加入”ss多核苷酸以便生成固定于所述固体基质上的ds多核苷酸。换句话说,所述条件被变成允许条件。然后可以将双链多核苷酸变性,以生成固定的ss多核苷酸及水相的ss多核苷酸。因此,本发明可扩展至一种生成以双链多核苷酸标记的固体基质或固体基质组合物的方法,所述方法包括:使用在第一组退火条件下具有相似退火特性但在第二组退火条件下具有差异退火特性的正向及反向引物进行双链的靶多核苷酸或其单链的衍生物的指数扩增;将扩增的扩增子产物与其上固定有至少一种这样的引物的固体基质或固体基质组合物接触,即该引物能够在第二组退火条件下与扩增子产物的链退火;并且将退火条件转换成所述第二组条件,从而有利于基于单引物的扩增以生成固定于所述固体基质上的单链的多核苷酸;将退火条件转换成第一组退火条件,借此在存在另一条引物的情况下,该单链的固定多核苷酸生成完整链或与所述固定链杂交的链以生成双链体多核苷酸标记的固体基质。在又一实施方案中,提供了一种方法用于生成以双链多核苷酸标记的固体基质或固体基质组合物,所述方法包括:在实现固定引物延伸的反应条件下,将其上具有以连接子方式结合的引物的固相载体与包含至少一种核酸分子的样品接触,其中所述固定引物选自:(i)与水相引物有序列同一性但具有不同于固定引物的熔解温度的引物;和(ii)嵌套于两个水相引物之间的引物。在描述本发明时,以下术语和内容被定义或者被阐明。本文提及的所有科学引文、专利、专利申请及制造商的技术说明书都通过引用的方式全文纳入本文。应理解的是,除非另外指出,否则本发明不限于具体的试剂、方法步骤或应用等,这是因为这些可以有变化。还应理解的是,本文使用的术语仅是为了描述具体实施方案的目的并且不是意欲进行限制。除非上下文中明确地声明相反,否则本说明书中使用的单数形式的“一”“一种”和“该”包括复数的方面。因此,例如提及“一个靶标”包括单个的靶标,以及两个或多个靶标;提及“一次扩增”包括单次扩增,以及多次扩增步骤;提及“该扩增子”包括单个的或多个的或复合体的扩增子;等等。本文使用的核酸化学、生物化学、遗传学和分子生物学的术语及符号遵循该领域中的那些标准的论文及教科书,例如kornbergandbaker,dnareplication,secondedition(w.h.freeman,newyork,1992);lehninger,biochemistry,secondedition(worthpublishers,newyork,1975);strachanandread,humanmoleculargenetics,secondedition(wiley-liss,newyork,1999);eckstein(ed),oligonucleotidesandanalogs:apracticalapproach(oxforduniversitypress,newyork,1991);gait(ed),oligonucleotidesynthesis:apracticalapproach(irlpress,oxford,1984);等等。“扩增子”是指多核苷酸扩增反应的产物。即,它是从一个或多个起始序列复制的一组多核苷酸,通常但不必需是双链的。所述一个或多个起始序列可以是相同序列的一个或多个拷贝,或者它可以是不同序列的混合物。扩增子的产生可以是通过其产物为一种或多种靶核酸的多复制物的多种扩增反应。一般而言,产生扩增子的扩增反应是“模板驱动的”,原因是反应物(核苷酸或寡核苷酸)的碱基配对在用于形成反应产物所需的模板多核苷酸中有互补物。在一个方面,模板驱动的反应是用核酸聚合酶的引物延伸,或者用核酸连接酶的寡核苷酸连接。这些反应包括但不限于pcr、线性聚合酶反应、nasba、滚环扩增等,它们被公开于通过引用的方式纳入本文的以下参考文献:mullisetal,美国专利4,683,195、4,965,188、4,683,202、4,800,159(pcr);gelfandetal,美国专利5,210,015(使用“taqman”探针的实时pcr);wittweretal,美国专利6,174,670;kacianetal,美国专利5,399,491(“nasba”);lizardi,美国专利5,854,033;aonoetal,日本专利公开jp4-262799(滚环扩增);等等。扩增反应可以是“实时”扩增,其中检测化学使得反应产物随扩增反应的进行而被测量。所述扩增可以为非对称扩增或对称扩增。本文使用的术语“扩增”是指进行扩增反应。“反应混合物”或“反应器”是指包含进行反应的所有必需反应物的溶液或容器,所述反应物包括但不限于在反应过程中将ph保持在选定水平的缓冲剂、盐、辅因子、清除剂等。“互补或基本上互补”是指在核苷酸或核酸之间的杂交、碱基配对或双链体的形成,例如在dsdna分子的两条链之间或者在寡核苷酸引物与单链核酸上的引物结合位点之间。互补的核苷酸通常为a与t(或a与u)或者c与g。在以下情形下两个单链的rna或dna分子被认为是基本互补的:即当一条链的核苷酸与另一条链的核苷酸(最佳地比对、比较并具有合适的核苷酸插入或删除)的配对至少为约80%时,通常至少约90%-95%,并且更优选地约98%-100%。或者,如果dna链例如在选择性杂交条件下与其互补物杂交,那么基本互补是存在的。一般地,在以下情形下会出现选择性杂交:当在具有至少14至25个核苷酸的一段上有至少约65%的互补时,优选至少75%,更优选至少约90%的互补。见kanehisanucleicacidsres.12:203,1984,其被通过引用的方式纳入本文。“双链体”是指至少两条这样的寡核苷酸及/或多核苷酸,即它们的核苷酸的所有的或大多数按照watson-crick型碱基配对完全地或部分地互补使得形成稳定的复合体。术语“退火”及“杂交”可互换使用,是指形成稳定的双链体。在言及双链体时“完全匹配的”是指,组成双链体的多核苷酸链或寡核苷酸链两者之间形成双链结构,使得每条链中的每个核苷酸与另一条链中的核苷酸进行watson-crick碱基配对。在言及基因组或靶多核苷酸时“遗传位点”或“位点”是指,基因组或靶多核苷酸的邻近的亚区域或片段。本文中使用的遗传位点或位点可以指基因组中的基因位置或基因部分,或者它可以指基因组序列的任意邻近部分,而不管它是否位于基因内或者是否与基因相关联。优选地,遗传位点是指从数十个核苷酸(例如长度10-30或10-100),至数百个核苷酸(例如长度100-1000或100-500),至数千个核苷酸(例如长度1000-10,000或1000-3000)的基因组序列的任意部分。在一些上下文中,遗传位点可以指核苷酸在基因组中的位置。“试剂盒”是指用于送递用于进行本发明的方法的材料或试剂的任何送递体系。在反应测定的上下文中,这种送递体系包括使得可以从一个位置至另一个位置保存、运输或送递反应试剂(例如在合适的容器中的探针、酶等)及/或支持材料(例如缓冲物、用于实施测定的书面说明书等)。例如,试剂盒包括包含相关反应试剂及/或支持材料的一个或多个外壳(例如盒子)。这些内含物可以被一起或分别地送递至目标受体。例如,第一容器可包含在测定中使用的酶,而第二容器包含探针。这些试剂盒还可以包含适宜于包含所述试剂的容器。在一个实例中,一个容器包含具有固定于其上的寡核苷酸、引物或多核苷酸的固体基质,所述寡核苷酸、引物或多核苷酸参与扩增反应。固体基质的一个实例为微阵列。因此,试剂盒可以是这样的整个扩增体系的一部分,即该扩增体系具有试剂组件、核酸组件、硬件组件和说明组件。言及“固体基质”包括用于进行扩增反应的任意结构形式。因此,例如乳液pcr被看作一种形式的固体基质,其中在不同相的乳液中进行pcr。“微阵列”是指具有平坦表面的固相载体,所述表面携带核酸阵列,阵列的每个成员均包含固定于空间确定区域或位点上的寡核苷酸或多核苷酸的相同拷贝,所述区域或位点不与阵列其他成员的那些区域或位点重叠;即,这些区域或位点是空间上不连续的。空间确定的杂交位点可以另外被“寻址”,这是因为其位置及其固定的寡核苷酸的身份在例如使用它之前是已知的或预先确定的。一般地,所述寡核苷酸或多核苷酸是单链的,并且通常通过5’端或3’端共价地连接于固相载体上。微阵列上包含核酸的非重叠区的密度通常每cm2超过100,更优选每cm2超过1000。微阵列技术在通过引用纳入的以下参考文献中被公开:schena(ed),microarrays:apracticalapproach(irlpress,oxford,2000);southern,currentopin.chem.biol.,2:404-410,1998。“随机微阵列”是指这样的微阵列,即其寡核苷酸或多核苷酸的空间上不连续区域不是空间上可寻址的。即,至少在开始时,所连接的寡核苷酸或多核苷酸的身份从其位置是不可辨别的。在一个方面,随机微阵列是微珠的平面阵列,其中每个微珠连接单一类型的杂交标签互补物,所述互补物例如来自最低限度相互杂交的一组寡核苷酸。同样地,在形成之后,随机阵列上的微珠或其寡核苷酸可以多种方式确定,包括通过光标记物例如荧光染料比率或量子点、形状、序列分析等。本文使用的“核苷”包括天然核苷,包括2’-脱氧形式及t-羟基形式,例如如kornbergandbaker,dnareplication,2nded.(freeman,sanfrancisco,1992)中所述。“聚合酶链反应”或“pcr”是指这样的反应,即通过dna互补链的同时引物延伸体外扩增特定的dna序列。换句话说,pcr反应可用于制备侧序列为引物结合位点的靶核酸的多个拷贝或复制物,这种反应包括以下步骤的一次或多次重复:(i)将靶核酸变性;(ii)将引物退火至所述引物结合位点,并且(iii)在存在三磷酸核苷的情况下通过核酸聚合酶延伸所述引物。通常地,所述反应在热循环仪器中循环经历为每一步骤最佳化的不同温度。每一步骤的具体温度、持续时间以及步骤之间变化的速率依赖于本领域普通技术人员熟知的多种因素,例如以下参考文献示例的:mcphersonetal.(eds),pcr:apracticalapproachandpcr2:apracticalapproach(irlpress,oxford,分别于1991年和1995年)。例如,在使用taqdna聚合酶的常规pcr中,可以将双链的靶核酸在>90℃的温度下变性,将引物在35-90℃的温度范围退火。术语“pcr”涵盖该反应的衍生形式,包括但不限于rt-pcr、实时pcr、嵌套pcr、定量pcr、多重pcr等。反应体积从数百纳升例如200nl至数百μl例如200μl。“逆转录pcr”或“rt-pcr”是指通过逆转录反应进行的pcr,所述pcr将靶rna转换成互补的单链dna,然后该单链dna被扩增,例如tecottetal,美国专利5,168,038,该专利通过引用的方式纳入本文。“实时pcr”是指随反应进行监测其反应产物即扩增子的量的pcr。有多种形式的实时pcr,区别主要在于用于监测反应产物的检测化学,例如gelfandetal,美国专利5,210,015(“taqman”);wittweretal,美国专利6,174,670和6,569,627(增补染料);tyagietal,美国专利5,925,517(分子信标(molecularbeacons));这些专利通过引用的方式纳入本文。实时pcr的检测化学被综述于mackayetal,nucleicacidsresearch,30:1292-1305,2002,该文献也通过引用的方式纳入本文。“嵌套pcr”是指一种两阶段pcr,其中第一次pcr的扩增子变成使用一组新引物的第二次pcr的样品,至少一个所述新引物可与所述第一扩增子的内部位置结合。“多核苷酸”或“寡核苷酸”可互换使用,均是指核苷酸单体的线性聚合物。组成多核苷酸和寡核苷酸的单体能够通过常规的单体-单体相互作用模式特异地结合成天然多核苷酸,所述单体-单体相互作用例如watson-crick型碱基配对、碱基堆积、hoogsteen碱基配对、反hoogsteen碱基配对等。在多核苷酸或寡核苷酸通过字母序列(大写字母或小写字母)例如“atgcctg”表示的任何时候,应理解的是除非在上下文中另外指出或是显而易见的,否则核苷酸从左至右的顺序为5’至3’,并且“a”表示脱氧腺苷、“c”表示脱氧胞苷、“g”表示脱氧鸟苷且“t”表示胸苷、“i”表示脱氧肌苷、“u”表示尿苷。“引物”为这样的天然的或合成的寡核苷酸,即其在与多核苷酸模板形成双链体时能够作为核酸合成的起始点,并沿模板从其3’端延伸使得形成延伸的双链体。引物的延伸通常是以核酸聚合酶例如dna聚合酶或rna聚合酶进行。在延伸过程中添加的核苷酸的序列是由模板多核苷酸的序列确定。引物通常利用dna聚合酶延伸。引物的长度范围通常从14个核苷酸至40个核苷酸,或者从18个核苷酸至36个核苷酸。引物可被应用于多种核酸扩增反应,例如使用单引物的线性扩增反应或者应用两个或多个引物的pcr。用于为特定应用的引物选择长度和序列的指导是本领域普通技术人员所公知的,这被通过引用方式纳入的以下参考文献证实:dieffenbach(ed),pcrprimer:alaboratorymanual,2ndedition(coldspringharborpress,newyork,2003)。“样品”是指来自生物、环境、医学或患者的一些材料,在其中进行靶核酸的检测或测量。在一方面,它是指包括试样或培养物(例如微生物培养物)。在另一方面,它是指同时包括生物样品和环境样品。样品可以包括合成来源的试样。生物样品可以是动物包括人的流体、固体(例如粪便)或组织,还可以是液体和固体的食物和饲料产品及拼料,例如乳制品、蔬菜、肉或肉副产物以及废料。生物样品可包括取自患者的材料,包括但不限于培养物、血液、唾液、脑脊液、胸腔液、乳、淋巴、痰、精液、穿刺抽出物等。生物样品可以获自所有不同科的家养动物,以及野生家畜(feralanimal)或野生动物,包括但不限于有蹄动物、熊、鱼、啮齿动物等这样的动物。环境样品包括环境材料例如表面物质、土壤、水和工业样品,以及来自食物和乳加工设备、器具、装置、器皿、一次性(disposable)产品和非一次性产品的样品。这些实例不能被解释为对本发明适用的样品类型的限制。“固体载体”、“载体”、“固相载体”和“固体基质”可互换使用,是指一种具有一个或多个硬或半硬表面的材料或一组材料。在许多实施方案中,所述固相载体的至少一个表面基本上应是平的,但是在一些实施方案中对于不同化合物可能需要具有例如孔、凸起区域、针、蚀刻沟等的物理上分开的合成区域。根据另一些实施方案,所述固相载体为珠子、树脂、凝胶、微球或其他几何构型的形式。如以上指出的,乳相被看作“固相载体”或“固体基质”。微阵列通常包含至少一种平面的固相载体,例如玻璃显微镜载玻片。所述载体还可以具有多个尺寸以使得可进行分拣。固相载体上的核酸分子/引物可以是被直接地固定或者通过化学键或核苷酸键固定。所有这种偶联化学被术语“连接子方式”涵盖。实施例1分步的非对称pcr方案图1(i)提供的示意表示为增强型固相pcr,用于将单链扩增子或双链扩增子加载至固相支持物上。待扩增的dna区域及来自其的扩增子以实线表示。虚线表示靶区域之外的序列。引物(以箭头表示)a表示“正向”pcr引物,b代表“反向”引物。在该实施例中,固相载体以圆圈表示。需要注意的是,固相载体缀合有多个拷贝的固相载体引物,但是为了示意图的简化仅给出了一条固相载体引物。波浪线表示非靶特异的连接子序列。较长、较宽的箭头表示较高靶特异性tm的引物。引物b可选地包含用于直接在检测体系中使用的标记分子,例如荧光(fluor)(以星号表示)。可以任选地包括标记分子作为反应底物,例如荧光标记的dntp。引物b任选地被设计,使得在给定温度范围内,引物b与靶序列的结合以及从该引物的聚合酶延伸的引发比引物a更有效。实现该目标的设计方法的一个实例包括的引物b具有比引物a更高的靶特异性熔解温度,使得在提高的退火温度下引物b结合并引发,而引物a不结合及引发。另一个实例为其中引物a在一特定温度下呈现“平锅柄抑制”,使得在该温度下引物b结合并引发,而引物a不结合及引发。引物a和引物b被应用为包括固相载体引物在内的常规pcr或非对称pcr的一部分。在允许所有引物的热循环条件下以包括引物a、引物b和固相载体缀合引物进行常规pcr。固相载体引物被设计,使得在给定的温度范围内从所述引物引发的聚合酶延伸比从引物a引发的聚合酶延伸更有竞争力。图1:(ii)2、(ii)3和(ii)4中描述的固相载体引物的设计,由于例如较高的tm及/或是被嵌套的或部分嵌套的,而提供了优于现有技术((ii)1)的改良的固相载体参与。现有技术(ii)1使用同一地匹配相应“水相”引物的靶特异性序列以及5-端连接子序列。在本文描述的实施例中,(ii)2包括3-端序列延伸,以相对于(ii)1提高tm;(ii)3包括这样的靶特异性序列,即该序列相对于(ii)1是嵌套的或部分嵌套的,但靶特异性tm类似于(ii)1;并且(ii)4包括这样的靶特异性序列,即该序列相对于(ii)1是嵌套的或部分嵌套的,且具有比(ii)1高的靶特异性tm。这些设计差异为固相载体引物参与提供了优于现有技术的动力学优点,使得有利于扩增子的固相载体加载。在后面的热循环过程中,退火步骤相对于前面的循环可以任选地具有升高的或降低的温度,使得固相载体引物仍退火以有效地靶向,但竞争者“水相”引物a不退火。例如,如果固相载体引物在升高的温度下呈现出比引物a更高的靶特异性tm,那么固相载体引物可引发但引物a不引发。另一个实施例为,引物a在低于一个特定温度的温度下呈现“平锅柄抑制”,使得低于该温度时固相载体引物结合并引发,而引物a不结合及引发。引物b可以被设计以具有与引物a或固相载体引物类似的引发特性。多次后面的这些循环的应用(其中引物b不能有效地结合靶)使得能够生成单链的固相载体扩增子。后面的这些循环的应用(其中引物b确实有效地结合靶)使得能够生成双链的固相载体扩增子。后面的这些循环的应用(其中引物b不能有效地结合靶)和随后的允许b结合的条件使得能够生成双链的固相载体扩增子。任选地,通过例如热变性或化学变性以及洗去溶液相或者例如外切核酸酶处理,可以在随后将双链的固相载体扩增子转换成单链的固相载体扩增子。实施例2固相分步的非对称pcr方案增强型固相pcr(esp-pcr)是本文设计的一种机制,该机制将高敏感的无损失对称“水相”pcr与有效的固相载体加载结合。esp-pcr通过通过消除“水相”引物和固相载体引物之间的竞争改变了将扩增子加载至固相载体上的机制,以增加固相载体引物的引发(图2)。“水相”引物不是必须被限制,因此使得体系更敏感。再者,esp-pcr固有的引物设计提供了这样的任选的可能性,即在专门允许固相载体引物结合的退火温度下进行后面的热循环。该实施例详细地描述了使用恒定固相载体材料、引物表面密度和“连接子”序列进行的esp-pcr,其范围和意图为剖析esp-pcr相对于sp-pcr的机制优点。寡核苷酸寡核苷酸购自integrateddnatechnologies(coralville,usa)。模板生成寡核苷酸以及esp-pcr和sp-pcr寡核苷酸在下文列出。缀合效率测量寡核苷酸(transprobe)序列为:/5ammc6/gtccatagctgtcctccct(seqidno:1)。所有寡核苷酸序列以5-端至3-端的方向列出。/5phos/、/5ammc6/、/5acryd/和/iammc6t/分别表示5-端磷酸、5-端胺修饰物、5-端亚磷酰胺(acrydite)和内部胺修饰物基团。ngo、ngps和cto3分别是指淋病奈瑟菌(neisseriagonorrhoea)opa、淋病奈瑟菌pils和沙眼衣原体(chlamydiatrachomatis)隐蔽性质粒orf3。下划线区域表示包括在固相载体引物的5-端部分中以有利于在“扩增子加载”过程中呈现靶特异性部分的非靶“连接子”序列。sp-pcr和esp-pcr固相载体引物共有的这种“连接子”序列还被用作transprobe杂交靶标,用于评估在缀合后至固相载体上的引物加载。寡核苷酸缀合于二氧化硅微球在使直径6.8μm的二氧化硅微球(bangslaboratories,fishers,usa)与寡核苷酸缀合之前将其经巯基(sulphydryl)功能化。整合dna技术产品的文献详细描述了固相载体硫醇(thiol)和5-端亚磷酰胺基团修饰的寡核苷酸之间的相互作用(可在线获自网站:http://scitools.idtdna.com/support/technical/technicalbulletinpdf/strategiesforattachingoligonucleotidestosolidsupports.pdf)。在缀合后,微球经一系列的5个离心/洗涤步骤处理以去除任何未结合的寡核苷酸,然后悬浮于缓冲液中至1mg/ml。通过以下方式评估寡核苷酸与微球的缀合水平:将1μl的均质微球悬液与5μl的含50mm氯化钠的缓冲液和0.5μl的10μmalexafluor647标记的transprobe(以比例为2:1的总transprobe:alexafluor647标记的transprobe)混合。alexafluor647购自molecularprobes(mountwaverley,australia)。使杂交混合物经过以下“杂交”热曲线:90℃下30秒,然后以1℃/10秒的速率冷却至20℃。在杂交后,在用facs阵列(bectondickinson,northryde,australia)分析前添加120μl缓冲液。本研究中呈现的数据来自该流水线的测定方案。微球可以在pcr后经受更严格的洗涤,其效应为降低“非模板对照”的背景荧光,同时不改变模板esp-pcr或sp-pcr样品的信号强度。将微球在以以下电压参数运行的同一台仪器上平行地评估3次:fcs550、ssc400、farred100、yellow650、nir200、red700、fcs阈值20000。*.fcs文件经fcsexpressv.3分析,以确定红色荧光的中值。student’st-检验被应用于证明esp-pcr和sp-pcr靶标对之间固相载体引物-微球结合相等。模板生成沙眼衣原体血清变型edna被用作使用cto3模板生成正向及反向引物的pcr的模板,以产生包括esp-pcr/sp-pcr寡核苷酸靶区域的扩增子。扩增子是使用qiaquick(注册)凝胶提取试剂盒(qiagen,doncaster,australia)纯化的琼脂糖胶。通过以hyperladderⅳdna标准品(bioline,alexandria,australia)为参照的产物凝胶电泳评估确定产率的大约值。类似地,淋病奈瑟菌atcc株43069被用作pcr模板,以使用它们各自的靶生成引物生成ngo和ngps靶标。esp-pcr和sp-pcr所有esp-pcr和sp-pcr均在使用1个单位hotstartaq[注册](qiagen,doncaster,australia)的20μl反应体积中进行。向hotstartaq(注册)反应缓冲液补充mg2+和dntp(newenglandbiolabs,genesearch,arundel,australia),以分别产生2mm和200μm的反应物浓度。在反应中包含有1μl的5μm“水相”正向引物、1μl的5μm“水相”反向引物(以2:1的总寡核苷酸:荧光-标记的寡核苷酸比率用alexafluor647标记)和1μl的1mg/ml固相载体引物缀合的微球的悬液。缀合sp-pcr或esp-pcr固相载体引物的微球分别被包括在sp-pcr和esp-pcr反应中。esp-pcr和sp-pcr为使用同一种主混合物(mastermix)平行地进行3次。在每种情况下,将引物和微球依照靶和模板进行分组。反应分别包含40000拷贝的cto3、40000拷贝的ngo和400000拷贝的ngps模板。应用的热曲线如下:94℃15分钟,然后[90℃30秒、44℃1分钟、72℃1分钟]的30个循环,然后[90℃、44℃2分钟、72℃2分钟]的5个循环。esp-pcr和sp-pcr的流式细胞分析在固相pcr后,将底部的5μl转移至96-孔微滴定板中的120μl的缓冲液中。*.fcs文件是以与前文所述相同的仪器设置通过facs阵列生成。使用fcsexpressv.3确定红色荧光图像的中值。凝胶电泳流式细胞术取样后,将8μl的残留pcr产物使用3%琼脂糖/tae凝胶以1.5μg的newenglandbiolabs100bp的梯状标准物(genesearch,arundel,australia)为参照进行分析。模板生成寡核苷酸cto3模板生成正向引物gatgcggaaaaagcttaccagcto3模板生成反向引物gggcttagaatcaccttctcgngo模板生成正向引物gcggattaacaaaaatcaggacaango模板生成反向引物taatctgccgctatcctccagngps模板生成正向引物ttttttgccggcgtggcatccngps模板生成反向引物atcgatatattatitccaccggaacesp-pcr和sp-pcr寡核苷酸通过esp-pcr的淋病奈瑟菌和沙眼衣原体检测的多重测定将以上列出的ngoesp-pcr和cto3esp-pcr固相载体引物分别与直径5.6μm和直径6.8μm的二氧化硅微球(bangslaboratories)缀合,洗涤并以每珠子群1mg/ml进行混合。1微升混合的珠子悬液被包含于下述esp-pcr反应中,所述esp-pcr反应包含淋病奈瑟菌opa引物:5’-ggcaacgmcgtaccggttt-3’(seqidno:20)和5-端alexafluor647-标记的5’-acgtcacagtttacgcgtttgaccggttaaaaaaagattttcac-3’(seqidno:21),以及沙眼衣原体隐蔽性质粒orf3引物:5’-agcttttaacaactttccaatcacta-3’(seqidi22)和5-端alexafluor647-标记的5’-acgactcactatagggtcccagagcttttgggtgtg-3’(seqidi23)。携带上面列出(见模板生成)涵盖沙眼衣原体隐蔽性质粒orf3的扩增子区域的淋病奈瑟菌atcc株43069基因组dna或质粒被用作pcr模板(包含5ngjurkat人基因组dna),作为对比的是仅人基因组dna或水对照。反应是以使用2个单位platinumtaq[商标](invitrogen)的20μl总体积进行,使用了以下的循环参数:94℃2分钟,然后[90℃30秒,55℃1分钟,72℃1分钟]的50个循环,然后72℃5分钟。将二氧化硅微球以缓冲液离心/洗涤2次,然后使用上文列出的仪器设置进行facs阵列分析。在缀合后将微球彻底洗涤,然后悬浮于缓冲液中至1mg/ml。寡核苷酸与微球缀合的水平的评估是通过以下方式进行的:将alexafluor647(molecularprobes)标记的“transprobe”杂交至固相载体引物连接子序列上,然后进行流式细胞分析。将微球在以以下电压参数运行的同一台仪器上平行地评估3次:fcs550、ssc400、farred100、yellow650、nir200、red700、fcs阈值20000。*.fcs文件经fcsexpressv.3分析,以确定红色荧光的中值。student’st-检验被应用于证明esp-pcr和sp-pcr靶对之间固相载体引物-微球缀合相等(ngopa,p=0.919;ngpils,p=0.759;ctcporf3,p=0.829)。照这样,与应用sp-pcr后相比,在应用esp-pcr后出现的扩增子加载方面的任何增加均应归因于改良的固相载体引发,而不能归因于不同的固相载体引物缀合水平。esp-pcr和sp-pcr之间的比较实验是在使用1个单位hotstartaq[注册](qiagen)的20μl反应体积中进行。向hotstartaq(注册)反应缓冲液补充mg2+和dntp(newenglandbiolabs),以分别产生2mm和200μm的反应物浓度。在反应中包含的有1μl的5μm“水相”正向引物、1μl的5μm“水相”反向引物(以2:1的总寡核苷酸:荧光-标记的寡核苷酸比率用alexafluor647标记)和1μl的1mg/ml固相载体引物缀合的微球的悬液。缀合sp-pcr或esp-pcr固相载体引物的微球分别被包括在sp-pcr和esp-pcr反应中。esp-pcr和sp-pcr为使用同一种主混合物平行地进行3次。在每种情况下,将引物和微球依照靶和模板(包含引物结合区域的线性dsdna)进行分组。反应分别包含40000拷贝的沙眼衣原体隐蔽性质粒orf3、40000拷贝的opa和400000拷贝的淋病奈瑟菌pils模板,或者无模板对照物(以显示背景荧光水平),并且应用以下的热曲线:94℃15min,然后[90℃30s,44℃1min,72℃1min]的30个循环,然后[90℃,44℃2min,72℃2min]的5个循环。在固相pcr后,将底部的5μl转移至96-孔微滴定板中的120μl的缓冲液中。*.fcs文件是以与前文所述相同的仪器设置通过facs阵列生成。使用fcsexpressv.3确定红色荧光图像的中值。esp-pcr在所有3个靶标评估中都产生比sp-pcr显著提高的固相载体扩增子加载(表3),有强的统计显著性:ngopa(9.89-倍,p=0.00000661)、ngpils(2.14-倍,p=0.001095)及ctcporf3(1.41-倍,p=0.000935)。p值与esp-pcr和sp-pcr之间扩增子加载无差异的零假设有关。倍数增加是指esp-pcrrfu/sp-pcrrfu。倍数增加在靶标之间的变化不是不可预知的,这是由于寡核苷酸之间杂交效率的内在差异以及其中包括复杂的竞争性杂交事件的事实。尽管有这种差异,esp-pcr在所研究的靶标中普遍是有利的。来自与用于流式细胞测量相同的反应器的非微球结合“水相”扩增子在esp-pcr后与在sp-pcr后,在所有靶之间有相同的产率,这是通过琼脂糖凝胶电泳评估的。在反应混合物中有或无微球时“水相”产物产率也是相同的,这表明二氧化硅微球与固相载体引物缀合不损害扩增效率。esp-pcr后,将固相载体经多重的离心及缓冲液交换步骤彻底洗涤,并用于包含前文使用的“水相”引物及“水相”形式的固相载体引物的模板受限的循环“再扩增”反应。大小与源自固相载体引物及“水相”反向引物的那些预期产物相符合的单产物带出现于凝胶电泳上,这表明esp-pcr固相支持物产物是特异的。总之,这些数据表明esp-pcr以一种特定方式成功地达到了这样的双重目标,即与标准sp-pcr相比不损害“水相”扩增并且提高扩增子的固相载体表面加载。这些出现的优点可归因于加载机制,其中:(i)固相载体引物的有效浓度由于其相对高的tm而被提高。指数后线性pcr(late-pcr)通过非对称pcr可提高单链产物的生成(sanchezetal,proc.natl.acad.sci.usa101:1933-1938,2004)。在该方法中,“更紧密结合的”、限制浓度的引物被应用以增加其在该反应中的“有效浓度”,这基于由“最近邻”公式所描述的引物tm和引物浓度之间的关系(santalucia,proc.natl.acad.sci.usa95:1460-1465,1998)。在esp-pcr中,该原理被应用于固相载体引物。“更紧密结合的”固相载体引物具有提高的“有效浓度”及改良的结合和引发动力学。(ii)固相载体引物相对于其“水相”对应物是嵌套的。因为固相载体引物可识别所述“水相”正向引物的不同结合位点,所以对于扩增子结合的直接竞争被消除。与sp-pcr不同,在esp-pcr中为了使固相载体引发被固相载体引物结合位点的“封闭”所抑制,“水相”正向引物必须结合于扩增子模板,聚合酶必须随后找到并结合该底物,并且聚合反应必须在短时间范围内紧接着进行。在标准sp-pcr和非对称sp-pcr二者中,水相引物与其引物结合位点的简单结合足以抑制固相载体引发。在pcr锁止(pcrclamping)的方法中,扩增可被内部的(嵌套的)肽核酸或锁核酸(lockednucleicacid)探针特异地阻断(dominguezandkolodney,oncogene24:6830-6834,2005;orumetal,nucleicacidsres.21:5332-5336,1993)。与esp-pcr的固相载体引物类似,pcr锁止方法的封闭探针可在引物结合并有机会发生聚合反应之前结合扩增子。仍然应用esp-pcr原理,使用“水相”引物及热循环参数,5个淋病奈瑟菌基因组和5个沙眼衣原体基因组等价物被每个单独的多重esp-pcr反应通过facs阵列检测。该方法使用缀合于不同直径的二氧化硅微球上的固相载体引物,以利于通过流式细胞术进行分辨。反应以包含沙眼衣原体靶区域的淋病奈瑟菌基因组dna和质粒为模板(包含jurkat人基因组dna),作为对比的是仅jurkat人基因组dna或水。表3固相载体扩增子加载与sp-pcr与sp-pcr相比,在esp-pcr后具有红色荧光标记的扩增子的二氧化硅微球的加载增加。rfu是指相对的荧光单位。括号中的数字代表3次重复平均值的标准误差。与sp-pcr比较,esp-pcr在以下的临床相关靶点中应用二氧化硅微球作为固相载体:淋病奈瑟菌opa(ngopa)、pils(ngpils)和沙眼衣原体隐蔽性质粒for3(ctcporf3)[mcleodetal,infectionandimmunity67:3469-3480,1999;comanduccietal,j.genmicrobiol139:1083-1092,1993]。简言之,esp-pcr和sp-pcr固相载体引物的制备如下:在使直径6.8μm的二氧化硅微球(bangslaboratories)与亚磷酰胺基团修饰的寡核苷酸缀合之前,对其进行巯基官能化。实施例3“热分步的”或“非分步的”esp-pcr和sp-pcr针对沙眼衣原体隐蔽性质粒orf3靶,平行地建立热分步的或非分步的方案作为实施例2中所述实验的一部分。对于“非分步的”pcr,采用热曲线44-44:94℃15min,然后[30个循环:90℃30秒、44℃1分钟、72℃1分钟],然后[5个循环:90℃、44℃2分钟、72℃2分钟]。对于“分步的”pcr,采用热曲线44-60:94℃15min,然后[30个循环:90℃30秒、44℃1分钟、72℃1分钟],然后[5个循环:90℃、60℃2分钟72℃2分钟]。结果显示于表4中。在固相pcr后,将底部的5μl转移至96-孔微滴定板中的120μl的缓冲液中。*.fcs文件是以与实施例2中所述相同的仪器设置通过应用bdfacs阵列生成。使用fcsexpressv.3确定红色荧光图像的中值。流式细胞术取样后,将8μl的残留pcr产物使用3%w/v琼脂糖/tae凝胶以1.5μg的newenglandbiolabs100bp的梯状标准物为对照(genesearch,arundel,australia)进行电泳。研究了作为esp-pcr方案一部分的最佳“热步骤”的潜在优点(44-60)。与“非分步的”热esp-pcr方案(44-44)相比,在后面的pcr循环过程中较高的退火温度提高了沙眼衣原体隐蔽性质粒orf3扩增子的表面加载((增加1.15倍)(student’st-检验,结果相同的可能性:p=0.0124))。来自同一反应器的非微球结合扩增子的凝胶电泳图被通过流式细胞术进行评估。在所有情况下,观察到可比较的“水相”pcr产物产率则表明微球扩增子加载的差异不受“水相”pcr产物产率的影响。在反应混合物中包含有或者无微球的情况下,水相产物产率之间没有可辨别的不同。表4“热分步的”或“非分步的”esp-pcr和sp-pcr与“非分步的”esp-pcr相比,“热分步的”esp-pcr后,具有红色荧光标记的扩增子的二氧化硅微球的加载增加。ctorf3是指沙眼衣原体隐蔽性质粒orf3。rfu是指相对的荧光单位。括号中的数字代表3次重复平均值的标准误差。本领域技术人员应了解,本文所述的发明允许除了被明确描述的那些之外的变化及修改。应理解的是,本发明包括所有这种变化及修改。本发明还单独地或综合地包括本说明书中提及或指出的所有步骤、特征、组合物和化合物,以及任何两种或多种所述步骤或特征的任意及所有组合。参考文献mcphersonetal.(eds),pcr:apracticalapproachandpcr2:apracticalapproach,irlpress,oxford,1991mcphersonetal.(eds),pcr:apracticalapproachandpcr2:apracticalapproach,irlpress,oxford,1995mackayetal,nucleicacidsresearch,30:1292-1305,2002orumetal,nucleicacidsres.21:5332-5336,1993sanchezetal,proc.natl.acad.sci.usa101:1933-1938,2004santalucia,proc.natl.acad.sci.usa95:1460-1465,1998schena(ed),microarrays:apracticalapproach(irlpress,oxford,2000);southem,currentopin.chem.biol.,2:404-410,1998shchepinovetal,nuc.acids.res.25:4447-44541997strachanandread,humanmoleculargenetics,secondedition,wiley-liss,newyork,1999tecottetdl,u.s.patentno.5,168,038序列表<110>d.j.帕克(仅对美国申请人)k.f.波特(仅对美国申请人)z.卡恩(仅对美国申请人)<120>核酸分子的生成<130>30468980/ejh<150>au2007900508<151>2007-02-02<150>au2007904458<151>2007-08-17<160>23<170>patentinversion3.4<210>1<211>19<212>dna<213>人工<220><223>寡核苷酸(transprobe)<400>1gtccatagctgtcctccct19<210>2<211>21<212>dna<213>人工<220><223>cto3模板生成正向引物<400>2gatgcggaaaaagcttaccag21<210>3<211>21<212>dna<213>人工<220><223>cto3模板生成反向引物<400>3gggcttagaatcaccttctcg21<210>4<211>24<212>dna<213>人工<220><223>ngo模板生成正向引物<400>4gcggattaacaaaaatcaggacaa24<210>5<211>21<212>dna<213>人工<220><223>ngo模板生成反向引物<400>5taatctgccgctatcctccag21<210>6<211>21<212>dna<213>人工<220><223>ngps模板生成正向引物<400>6ttttttgccggcgtggcatcc21<210>7<211>25<212>dna<213>人工<220><223>ngps模板生成反向引物<400>7atcgatatattatttccaccggaac25<210>8<211>18<212>dna<213>人工<220><223>cto3”水相”正向引物<400>8acagacccttctctaggt18<210>9<211>43<212>dna<213>人工<220><223>cto3”水相”反向引物<400>9aattctaatacgactcactatagggcttttgggtgtgactgtg43<210>10<211>43<212>dna<213>人工<220><223>cto3sp-pcr固体支持物引物<400>10aattaaagggaggacagctatggacacagacccttctctaggt43<210>11<211>60<212>dna<213>人工<220><223>cto3esp-pcr固体支持物引物<400>11aattaaagggaggacagctatggaccactaataaaattcaatgcaacgggttattcactc60<210>12<211>20<212>dna<213>人工<220><223>ngo”水相”正向引物<400>12gccatattgtgttgaaacac20<210>13<211>46<212>dna<213>人工<220><223>ngo”水相”反向引物<400>13aattctaatacgactcactataggggtttgaccggttaaaaaaaga46<210>14<211>45<212>dna<213>人工<220><223>ngosp-pcr固体支持物引物<400>14aattaaagggaggacagctatggacgccatattgtgttgaaacac45<210>15<211>55<212>dna<213>人工<220><223>ngoesp-pcr固体支持物引物<400>15aattaaagggaggacagctatggaccccgatataatccgcccttcaacatcagtg55<210>16<211>19<212>dna<213>人工<220><223>ngps”水相”正向引物<400>16aatgaggcaaattaggcct19<210>17<211>45<212>dna<213>人工<220><223>ngps”水相”反向引物<400>17aattctaatacgactcactatagggcttgcaaacccttaaaagac45<210>18<211>44<212>dna<213>人工<220><223>ngpssp-pcr固体支持物引物<400>18aattaaagggaggacagctatggacaatgaggcaaattaggcct44<210>19<211>54<212>dna<213>人工<220><223>ngpsesp-pcr固体支持物引物<400>19aattaaagggaggacagctatggacaaatcaagcggtaagtgatttcccacggc54<210>20<211>19<212>dna<213>人工<220><223>淋病奈瑟菌opa引物<400>20ggcaacgmcgtaccggttt19<210>21<211>44<212>dna<213>人工<220><223>alexafluor647-标记的5'<400>21acgtcacagtttacgcgtttgaccggttaaaaaaagattttcac44<210>22<211>26<212>dna<213>人工<220><223>沙眼衣原体隐蔽性质粒orf3引物<400>22agcttttaacaactttccaatcacta26<210>23<211>36<212>dna<213>人工<220><223>alexafluor647-标记的5'<400>23acgactcactatagggtcccagagcttttgggtgtg36当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1
- 抗结核杆菌感染的小分子核苷酸dna适配子及其制备方法
- 一种用于促进vhl基因表达的分离的核苷酸分子和促进剂及其制备方法
- 核苷酸分子septin1sr2及其在制备治疗白血病药物中的应用的制作方法
- 核苷酸分子septin1sr1及其在制备免疫制剂中的应用的制作方法
- 核苷酸分子sr5b2及其在制备抗糖尿病药物中的应用的制作方法
- 核苷酸分子sr3b2及其在制备抗糖尿病药物中的应用的制作方法
- 核苷酸分子sr2b2及其在制备抗糖尿病药物中的应用的制作方法
- 核苷酸分子sr1b2及其在制备抗糖尿病药物中的应用的制作方法
- 核苷酸分子sr2b及其在制备抗糖尿病药物中的应用的制作方法
- 核苷酸分子sr4b及其在制备抗糖尿病药物中的应用的制作方法