一种硫代修饰引物及应用该引物的SNP检测方法与流程
本发明属于snp检测技术领域,特别涉及一种硫代修饰引物及应用该引物的snp检测方法。
背景技术:
单核苷酸多态性(singlenucleotidepolymorphism,snp),主要是指在基因组水平上由单个核苷酸的变异所引起的dna序列多态性。它是人类可遗传的变异中最常见的一种。占所有已知多态性的90%以上。snp在人类基因组中广泛存在,平均每500~1000个碱基对中就有1个,估计其总数可达300万个甚至更多。
目前snp的常用检测方法是等位基因特异性pcr(alleleapecificpcr,as-pcr),as-pcr是基于引物3`端碱基与dna模板匹配与否来区分snp位点。理论上,只要末位碱基不配对,那么引物无法有效延伸;但是末位单碱基错配往往不能达到预期的分辨效果。由于pcr反应中dna呈指数级增长,当与引物3`端碱基错配的dna模板在第一个循环有少量的扩增时,其复制出来的dna模板在后续的循环里与原引物的错配就不存在。随着指数级的dna扩增,错配产物的数量也相当可观。达到影响分辨率的程度。
授权公告号为cn103320514b的发明专利公开了一种检测临近snp的多重pcr方法,可以解决扩增抑制问题并提高扩增效率;但是该方法需要设计多对引物,工作量较大,操作较为繁琐。因此,提供一种新的snp检测方法,以提高灵敏性和准确性,防止错配延伸,成为本领域研究人员急需解决的技术问题。
技术实现要素:
为了解决上述技术问题,本发明提供了一种硫代修饰引物及应用该引物的snp检测方法。
本发明具体技术方案如下:
本发明一方面提供了一种硫代修饰引物,所述引物的硫代修饰位点为3`端第一和第二个碱基之间的磷酸二酯键。
本发明另一方面提供了一种应用该引物的的snp检测方法,包括如下步骤:
s1:设计并合成用于扩增目的基因的特异性引物,所述特异性引物包括两条上游引物f1、f2和一条下游引物r,所述f1引物和所述f2引物3`端第一位碱基不同,其余序列均相同;
s2:分别对所述f1引物和所述f2引物进行硫代修饰;
s3:用所述引物对目的基因进行pcr扩增;
s4:对实验结果进行分析。
进一步地,所述pcr的反应体系中使用的dna聚合酶为pfu高保真dna聚合酶。
snp位点的识别,即单个碱基的识别。高保真dna聚合酶比普通taqdna聚合酶对错配碱基的识别能力更强。但是高保真dna聚合酶具有3`-5`核酸外切酶活性,在pcr的过程中会切掉不匹配的碱基;硫代修饰的作用是改变碱基间的连接方式,将碱基间磷酸二酯键中的氧原子换成硫原子,3`-5`核酸外切酶在pcr过程中不能识别该位点,从而不能将不匹配的碱基切除。由此可以提高snp检测的特异性和准确性。
进一步地,所述步骤s3中,分别采用f1引物和f2引物为上游引物、以r引物为下游引物进行实时荧光定量pcr。
进一步地,所述pcr的反应体系中包括如下组分:
模板dna0.2ng/μl、f1引物或f2引物0.5μm、r引物0.5μm、dntp0.2mm、pfu高保真dna聚合酶0.02u/μl以及1×的sybrgreen。
不同种类生物的不同基因片段大小不同,差距可达两个数量级,用量约在0.02~2ng/μl之间。本研究中应用的是人基因组片段,因此模板用量为0.2ng/μl。
进一步地,所述pcr的反应条件如下:
(1)预变性:98℃3min;
(2)扩增:98℃10s,58℃30s,共40个循环;
(3)扩增曲线:95℃15s;60℃15s;95℃15s。
分别以两种上游引物进行扩增,当特异性引物与模板完全匹配时,引物可以正常延伸,产生扩增曲线;当特异性引物与模板不能匹配时,引物不能延伸,不能产生扩增曲线。由此,可以对模板中的snp位点起到检测作用。
上述技术方案可以通过设计分子信标探针替代荧光染料,此时,所述步骤s3包括如下步骤:
s3.1:对目的基因通过as-pcr进行位点识别;
s3.2:分子信标扩增:利用两条分子信标探针对步骤s3.1中得到的扩增产物进行pcr扩增;
s3.3:信号产生:将反向引物结合到步骤s3.2中得到的扩增产物上,进行pcr扩增,使分子信标的荧光基团和淬灭基团分开,从而产生信号。
第一轮pcr用于识别等位基因,产生大量带有等位基因通用标签的pcr产物;第二轮pcr是以分子信标探针作为引物,结合到通用标签序列部分,进行pcr扩增;第三轮pcr通过反向引物扩增,将分子信标的发夹结构打开,使分子信标的荧光基团和淬灭基团分开,从而产生信号。
进一步地,所述f1引物的5`端连接有通用标签1,所述f2引物的5`端连接有通用标签2,所述通用标签1和所述通用标签2的碱基序列如下:
通用标签1:5`gaaggtgaccaagttcatgct3`;
通用标签2:5`gaaggtcggagtcaacggatt3`。
进一步地,两条所述分子信标探针的3`端引物序列分别与通用标签1和通用标签2的序列相同,5`端分别标记fam和hex作为荧光基团,中间均标记dabcyl作为淬灭基团。
进一步地,所述步骤s3中pcr反应体系中包括如下成分:
模板dna0.2ng/μl、f1引物0.5μm、f2引物0.5μm、r引物0.5μm、分子信标beacon-fam0.5μm、分子信标beacon-hex0.5um、dntp0.2mm以及taqdna聚合酶0.02u/μl;
所述pcr的反应条件如下:
(1)预变性:94℃5min;
(2)扩增:94℃20s,58℃30s,共36个循环。
将三轮pcr所需的试剂合在一个反应体系中,通过上述反应条件,即可通过一次pcr达到三轮pcr的效果。
本发明的有益效果如下:本发明提供了一种硫代修饰引物及应用该引物的snp检测方法,pcr过程中采用高保真dna聚合酶,提高了对特异性引物的识别能力,同时对引物进行硫代修饰,避免了高保真酶将不匹配的碱基切下来,提高检测方法的特异性;实时定量检测中,通过扩增曲线可以判定是否有非特异性扩增,利用△ct判定分型结果,可以避免终点定量因非特异性扩增造成假阳性的问题,具有较高的准确度和灵敏性;该方法还可以借助分子信标探针替代核酸染料,并将三步pcr扩增合为一步反应,以此达到同样的效果,可以提高该方法的可行性,为实验操作提供了多种选择。
附图说明
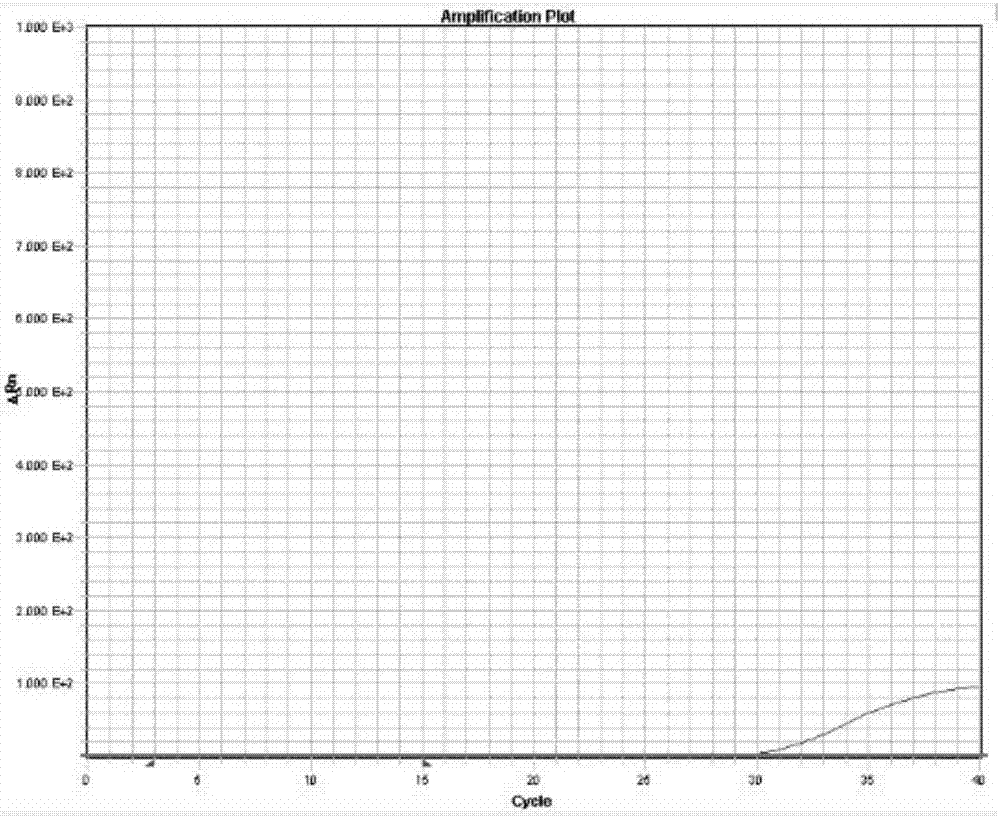
图1为实验例1中扩增曲线1的示意图;
图2为实验例1中扩增曲线2的示意图;
图3为实验例1中扩增曲线1与扩增曲线2的对比情况;
图4为实验例2中扩增结果示意图。
具体实施方式
下面结合附图和以下实施例对本发明作进一步详细说明。
实施例1
一种利用碱基错配扩增的snp检测方法,包括如下步骤:
s1:以人基因组snp位点rs642431为目的基因,设计并合成用于扩增目的基因的特异性引物,所述位点碱基序列如下:
所述引物的碱基序列如下:
f1:5`cggcacaccagtgtgtgtct*t3`;
r:5`attctgggaaccagggaagatgtgg3`;
其中f1引物经过硫代修饰,硫代修饰的位点为3`端倒数第一和倒数第二个碱基之间的磷酸二酯键;
s2:用所述引物对目的基因进行pcr扩增,具体反应体系如下;
模板dna10ng、f1引物0.5μm、r引物0.5μm、dntp0.2mm、pfu高保真dna聚合酶1u以及2.5μl的sybrgreen(20×),补充ddh2o至终体积50μl;
反应条件如下:
(1)预变性:98℃3min;
(2)扩增:98℃10s,58℃30s,共40个循环;
(3)扩增曲线:95℃15s;60℃15s;95℃15s;
s3:对实验结果进行分析。
实施例2
一种利用碱基错配扩增的snp检测方法,包括如下步骤:
s1:以人基因组snp位点rs642431为目的基因(碱基序列同实施例1),设计并合成用于扩增目的基因的特异性引物,所述引物的碱基序列如下:
f2:5`cggcacaccagtgtgtgtct*g3`;
r:5`attctgggaaccagggaagatgtgg3`;
其中f2引物经过硫代修饰,硫代修饰的位点为3`端倒数第一和倒数第二个碱基之间的磷酸二酯键;
s2:用所述引物对目的基因进行pcr扩增,具体反应体系如下;
模板dna10ng、f2引物0.5μm、r引物0.5μm、dntp0.2mm、pfu高保真dna聚合酶1u以及2.5μl的sybrgreen(20×),补充ddh2o至终体积50μl;
反应条件如下:
(1)预变性:98℃3min;
(2)扩增:98℃10s,58℃30s,共40个循环;
(3)扩增曲线:95℃15s;60℃15s;95℃15s;
s3:对实验结果进行分析。
实施例3
一种利用碱基错配扩增的snp检测方法,包括如下步骤:
s1:以人基因组snp位点rs645112为目的基因,设计并合成用于扩增目的基因的特异性引物和分子信标探针,所述位点碱基续列如下:
其中f1引物和f2引物的5`端分别连接有通用标签1和通用标签2,连接有通用标签的引物序列如下:
f1`:5`gaaggtgaccaagttcatgcttggaaaacagaagcactaagttctt3`;
f2`:5`gaaggtcggagtcaacggatttggaaaacagaagcactaagttctg3`。
r:5`cacaaagatgatagaaattaagatttcaatggc3`;
两条分子信标探针的3`端引物序列分别与通用标签1和通用标签2的序列相同,所述分子信标探针的碱基序列如下:
beacon-fam:fam-5`acgcgaagttaagacctatgcgcgt3`(dabcyl)gaaggtgaccaagttcatgct-磷酸化;
beacon-hex:hex-5`acgcgaagttaagacctatgcgcgt3`(dabcyl)gaaggtcggagtcaacggatt-磷酸化。
s2:用所述引物和分子信标探针对目的基因进行pcr扩增,具体反应体系如下;
模板dna10ng、f1引物0.5μm、f2引物0.5μm、r引物0.5μm、分子信标beacon-fam0.5μm、分子信标beacon-hex0.5μm、dntp0.2mm以及taqdna聚合酶1u,补充ddh2o至终体积50μl;
反应条件如下:
(1)预变性:94℃5min;
(2)扩增:94℃20s,58℃30s,共36个循环。
s3:对实验结果进行分析。
实验例1
提取目的基因,分别按照实施例1和实施例2中提供的方法对目的基因进行snp检测,并分别进行多个平行实验;以实施例1的多个平行实验生成扩增曲线1,以实施例2的多个平行实验生成扩增曲线2,并进行比较。
实验结果如图1~3所示,由于f1引物与模板能匹配,而f2引物与模板不能匹配,因此以f2作为上游引物的pcr扩增受到抑制,其扩增曲线的ct值比以f1为上游引物时更大,并且△ct>3,因此认为该位点为纯合。
实验例2
提取目的基因,按照实施例3中提供的方法对目的基因进行snp检测,并进行多次平行实验,对结果进行分析。实验结果如图4所示,靠近y轴的数据点最多,占所有数据点中的一半,靠近x轴的数据点和趋向坐标中间的数据点的数量接近。表明该snp位点上g的纯合子较多,t的纯合子以及g-t杂合子也有检出。
以上所述实施例仅表达了本发明的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对本发明专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。因此,本发明专利的保护范围应以所附权利要求为准。
- 还没有人留言评论。精彩留言会获得点赞!