丙炔氟草胺晶体的制备方法和丙炔氟草胺的制备方法与流程
本发明涉及有机合成领域,具体地,涉及a-晶型丙炔氟草胺的制备方法、a-晶型和2nd-晶型混合的丙炔氟草胺的制备方法以及丙炔氟草胺的制备方法。
背景技术
丙炔氟草胺(其他通用名:速收)是日本住友化学工业株式会社创制并开发的由幼芽和叶片吸收的n-苯基邻氨甲酞亚胺类除草剂,在土壤处理中可有效防除1年生阔叶杂草和部分禾本科杂草,在环境中易降解,对后茬作物安全。大豆、花生对其有很好的耐药性,玉米、小麦、大麦、水稻具有中等忍耐性,因此该除草剂在农业生产方面具有广泛的应用,并具备巨大的经济市场价值。
丙炔氟草胺的合成已有多种合成方法,例如us4640707中公开了通过6-氨基-7-氟-4-丙炔基-1,4-苯并恶嗪-3(4h)-酮与3,4,5,6-四氢苯酐和醋酸反应,可制得丙炔氟草胺。
刘安昌等对新型除草剂丙炔氟草胺的合成进行了研究(新型除草剂丙炔氟草胺的合成研究,《世界农药》,2011,33(2),27-29),其中,将6-氨基-4-(2-丙炔基)-2h-1,4-苯并恶嗪-3(4h)-酮、3,4,5,6-四氢苯酐和冰醋酸加热至回流,反应2h后终止,经过萃取分离、洗涤、浓缩冷却得到丙炔氟草胺晶体。
jph0597848公开了采用(1,3-二甲基丁烯氨基)-7-氟-4-(2-炔丙基)-1,4-苯并恶嗪-3(4h)-酮的甲基异丁基甲酮溶液与3,4,5,6-四氢苯酐反应生成丙炔氟草胺,收率为95%,含量为99%。
cn104169274a公开了将丙炔氟草胺在60℃下溶解在甲基异丁基酮中,以调节浓度至10.1mg/ml,将溶剂快速冷却至0℃,随后静置得到a-晶型丙炔氟草胺。然而该方法需要额外的冷却介质进行冷却结晶,结晶条件比较苛刻,操作复杂。
wo2013187491a中公开了使用不同的溶剂中制备出丙炔氟草胺的另外七种不同的晶型,分别为1st晶体、2nd晶体、3rd晶体、4th晶体、5th晶体,6th晶体和7th晶体。其中,将丙炔氟草胺在60℃下溶解在四氢呋喃中,以调节浓度为51.0mg/ml,将得到的混合物逐渐滴加到加热至100℃的玻璃板上以快速除掉其中的溶剂,从而得到2nd晶体。然而该方法的操作比较复杂。
尽管现有技术中已经能够制备a-晶型丙炔氟草胺,但操作总体比较复杂。
技术实现要素:
本发明的目的是克服现有制备a-晶型丙炔氟草胺的方法存在操作复杂的缺陷,提供新的a-晶型丙炔氟草胺的制备方法,a-晶型和2nd-晶型混合的丙炔氟草胺的制备方法以及丙炔氟草胺的制备方法。
根据本发明的第一方面,本发明提供一种a-晶型丙炔氟草胺的制备方法,该方法包括:将处于第一温度的溶有丙炔氟草胺的热溶液降温至结晶温度进行结晶,得到a-晶型丙炔氟草胺,其中,结晶温度为80-100℃,第一温度高于结晶温度,所述热溶液的溶剂为ch3cooh和/或单烷基苯,所述单烷基苯中的烷基为c1-c3的烷基。
根据本发明的第二方面,本发明还提供了一种a-晶型丙炔氟草胺的制备方法,该方法包括:将丙炔氟草胺在加热状态下溶解于溶剂中,在保持加热状态下,向所得热溶液中加入水进行结晶得到a-晶型丙炔氟草胺,其中,加热的温度为20-100℃,所述溶剂为四氢呋喃、n,n-二甲基甲酰胺和丙酮中的一种或多种。
根据本发明的第三方面,本发明还提供了一种a-晶型和2nd-晶型混合的丙炔氟草胺的制备方法,该方法包括:将处于第二温度的溶有丙炔氟草胺的热溶液降温至结晶温度进行结晶,得到a-晶型和2nd-晶型混合的丙炔氟草胺,其中,结晶温度为50-65℃,第二温度高于结晶温度,所述热溶液的溶剂为ch3cooh和/或单烷基苯,所述单烷基苯中的烷基为c1-c3的烷基。
根据本发明的第四方面,本发明提供一种丙炔氟草胺的制备方法,该方法包括:在催化剂的存在下,将式(1)所示的6-氨基-7-氟-4-丙炔基-1,4-苯并恶嗪-3(4h)-酮与式(2)所示的3,4,5,6-四氢苯酐在第一溶剂中接触反应,所述催化剂为碱性含氮有机物、或者有机酸和碱性含氮有机物的混合物,其中,所述方法还包括采用上述方法将接触反应得到的丙炔氟草胺进行纯化,
根据本发明的第五方面,本发明还提供了一种丙炔氟草胺的制备方法,该方法包括:将式(1)所示的6-氨基-7-氟-4-丙炔基-1,4-苯并恶嗪-3(4h)-酮与式(2)所示的3,4,5,6-四氢苯酐在第二溶剂中接触反应,所述第二溶剂为ch3cooh,其中,所述方法还包括采用上述方法将接触反应得到的丙炔氟草胺进行纯化,
本发明采用特定的溶剂和特定的结晶温度制备a-晶型丙炔氟草胺,操作简单,利于工业化生产。
本发明采用特定的溶剂和特定的结晶温度制备a-晶型和2nd-晶型混合的丙炔氟草胺,操作简单,利于工业化生产。
本发明的其它特征和优点将在随后的具体实施方式部分予以详细说明。
附图说明
附图是用来提供对本发明的进一步理解,并且构成说明书的一部分,与下面的具体实施方式一起用于解释本发明,但并不构成对本发明的限制。在附图中:
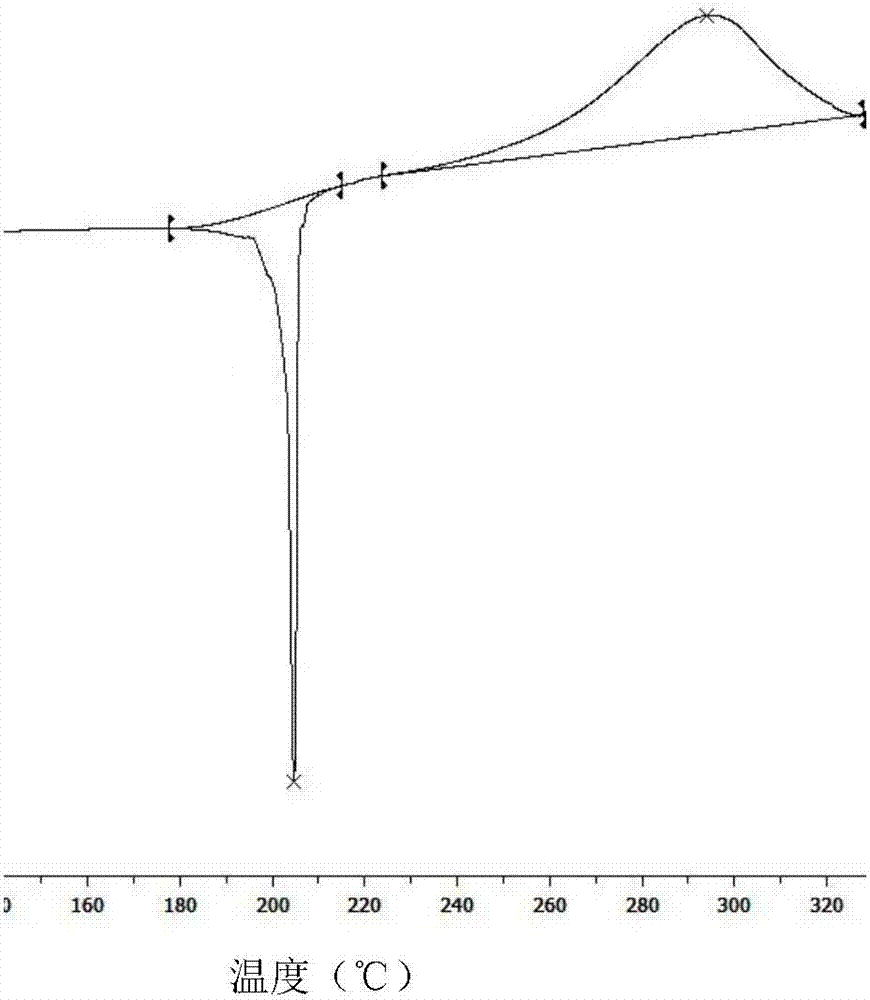
图1是由实施例1得到的a-晶型丙炔氟草胺的dsc图。
图2是由实施例1得到的a-晶型丙炔氟草胺的x射线衍射谱图。
图3是由实施例13得到的a-晶型和2nd-晶型混合的丙炔氟草胺的dsc图。
图4是由实施例13得到的a-晶型和2nd-晶型混合的丙炔氟草胺的x射线衍射谱图。
具体实施方式
以下对本发明的具体实施方式进行详细说明。应当理解的是,此处所描述的具体实施方式仅用于说明和解释本发明,并不用于限制本发明。
在本文中所披露的范围的端点和任何值都不限于该精确的范围或值,这些范围或值应当理解为包含接近这些范围或值的值。对于数值范围来说,各个范围的端点值之间、各个范围的端点值和单独的点值之间,以及单独的点值之间可以彼此组合而得到一个或多个新的数值范围,这些数值范围应被视为在本文中具体公开。
根据本发明的第一方面,本发明提供了一种a-晶型丙炔氟草胺的制备方法,该方法包括:将处于第一温度的溶有丙炔氟草胺的热溶液降温至结晶温度进行结晶,得到a-晶型丙炔氟草胺,其中,结晶温度为80-100℃,第一温度高于结晶温度,所述热溶液的溶剂为ch3cooh和/或单烷基苯,所述单烷基苯中的烷基为c1-c3的烷基。
在本发明中,ch3cooh优选以醋酸的形式提供,例如浓度为95-99重量%的醋酸,更优选为97-99重量%的醋酸,最优选为99重量%的醋酸。
根据本发明的方法,为了获得更高收率和更高纯度的a-晶型丙炔氟草胺,优选情况下,所述单烷基苯为甲苯和/或乙苯,更优选为甲苯。
本发明将处于第一温度的溶有丙炔氟草胺的热溶液降温至结晶温度进行结晶,第一温度只要能确保丙炔氟草胺在开始结晶前不析出即可,一般地,所述第一温度为90-120℃,优选情况下,所述热溶液的溶剂为ch3cooh,所述第一温度为95-110℃;所述热溶液的溶剂为单烷基苯,所述第一温度为95-110℃。
根据本发明的方法,优选情况下,所述结晶温度为80-90℃,在该温度下结晶速度较快,且获得的a-晶型丙炔氟草胺的收率和纯度较高。
根据本发明的方法,结晶的时间可以根据结晶温度进行合理的选择,一般地,所述结晶的持续时间可以为1-3小时。进一步优选地,所述结晶在伴随搅拌的状态下进行。
根据本发明的方法,所述溶剂的用量以能够实现降温结晶的目的为准,一般地,相对于1g的丙炔氟草胺,所述溶剂的用量为1-20ml,为了获得更高收率和更高纯度的a-晶型丙炔氟草胺,优选情况下,相对于1g的丙炔氟草胺,所述溶剂的用量为1-11ml。
本发明的方法还包括将结晶之后所得混合物降温至环境温度进行固液分离的步骤,以将a-晶型丙炔氟草胺晶体分离出来。所述固液分离的方法可以为本领域所公知的各种常规的固液分离方法,例如可以为过滤、抽滤、压滤、离心分离等。优选情况下,所述方法还包括将经固液分离得到的固体进行洗涤和干燥。其中,所述环境温度可以为10-30℃。
根据本发明的方法,所述热溶液可以通过以下方法制备:将丙炔氟草胺溶解于溶剂中,可选地,将部分溶剂去除。所述溶解的温度可以根据溶剂的种类进行合理的选择,一般地,所述溶解的温度可以为90-120℃。优选情况下,所述溶剂为ch3cooh,所述溶解的温度为95-110℃;溶解优选在加热回流的状态下进行,所述加热回流的时间可以为0.1-5小时,优选为0.5-2小时。所述溶剂为单烷基苯,且所述溶解的温度为95-110℃;溶解优选在加热回流的状态下进行,所述加热回流的时间可以为0.1-5小时,优选为0.5-2小时。为了加快溶解,所述溶解优选在搅拌状态下进行。
根据本发明的方法,可以根据热溶液中的溶剂的用量,将所述热溶液在降温至结晶温度之前进行去除部分溶剂的步骤,以提高a-晶型丙炔氟草胺的收率和纯度。去除溶剂的方法可以为本领域中常用的方法,例如常压蒸馏,一般地,所述溶剂为ch3cooh,常压蒸馏的温度可以为115-130℃;所述溶剂为单烷基苯时,常压蒸馏的温度可以为108-115℃。
根据本发明的方法,所述丙炔氟草胺可以通过商购得到,也可以通过本领域中常用的合成方法进行制备。
根据本发明的第二方面,本发明还提供了一种a-晶型丙炔氟草胺的制备方法,该方法包括:将丙炔氟草胺在加热状态下溶解于溶剂中,在保持加热状态下,向所得热溶液中加入水进行结晶得到a-晶型丙炔氟草胺,其中,加热的温度为20-100℃,所述溶剂为四氢呋喃、n,n-二甲基甲酰胺和丙酮中的一种或多种。
本发明中,所述加热的温度以确保在进行结晶前丙炔氟草胺不会析出,优选情况下,所述热溶液的溶剂为四氢呋喃,所述加热的温度为60-70℃;所述加热优选在回流的状态下进行,加热回流的时间可以为0.1-5小时,优选为0.5-2小时。所述热溶液的溶剂为n,n-二甲基甲酰胺,所述加热的温度为20-100℃;所述加热优选在回流的状态下进行,加热回流的时间可以为0.1-5小时,优选为0.5-2小时。所述热溶液的溶剂为丙酮,所述加热的温度为60-70℃。所述加热优选在回流的状态下进行,加热回流的时间可以为0.1-5小时,优选为0.5-2小时。为了加快溶解,所述溶解优选在搅拌状态下进行。
根据本发明的方法,在加热状态下向所得热溶液中加入水即可实现结晶得到a-晶型丙炔氟草胺,一般情况下,结晶的持续时间为1-3小时。所述结晶优选在伴随搅拌的状态下进行。
根据本发明的方法,所述溶剂的用量以能够将丙炔氟草胺完全溶解为准,一般地,相对于1g的丙炔氟草胺,所述溶剂的用量可以为1-20g,为了获得更高收率和更高纯度的a-晶型丙炔氟草胺,优选情况下,相对于1g的丙炔氟草胺,所述溶剂的用量可以为5-15g。
本发明中,所述水的用量以能够使丙炔氟草胺析晶为准。一般地,相对于1g的丙炔氟草胺,所述水的用量可以为5-30ml,优选为20-25ml。
本发明中,所述水的温度以能够将a-晶型丙炔氟草胺从热溶液中结晶析出为准,一般地,所述水的温度可以为10-25℃。所述水的种类可以为本领域中常用的水,例如自来水、蒸馏水和去离子水中的一种或多种。
本发明的方法还包括将结晶之后所得混合物降温至环境温度进行固液分离的步骤,以将a-晶型丙炔氟草胺晶体分离出来。所述固液分离与上文描述一致,在此不再赘述。优选情况下,所述方法还包括将经固液分离得到的固体进行洗涤和干燥。其中,所述环境温度可以为10-30℃。
根据本发明的方法,根据热溶液的溶剂的用量,所述方法可以包括将丙炔氟草胺在加热状态下溶解于溶剂中,在保持加热状态下,向所得热溶液中加入水后将部分溶剂去除的步骤,以提高a-晶型丙炔氟草胺的收率和纯度。去除溶剂的方法可以为本领域中常用的方法,例如常压蒸馏,一般地,所述溶剂为四氢呋喃,常压蒸馏的温度可以为60-70℃;所述溶剂为丙酮,常压蒸馏的温度可以为60-70℃;所述溶剂为n,n-二甲基甲酰胺,常压蒸馏的温度可以为20-100℃。
根据本发明的方法,所述丙炔氟草胺可以通过商购得到,也可以通过本领域中常用的合成方法进行制备。
根据本发明的第三方面,本发明还提供了一种a-晶型和2nd-晶型混合的丙炔氟草胺的制备方法,该方法包括:将处于第二温度的溶有丙炔氟草胺的热溶液降温至结晶温度进行结晶,得到a-晶型和2nd-晶型混合的丙炔氟草胺,其中,结晶温度为50-65℃,第二温度高于结晶温度,所述热溶液的溶剂为ch3cooh和/或单烷基苯,所述单烷基苯中的烷基为c1-c3的烷基。
本发明中,“第一温度”和“第二温度”没有特别的含义,分别表示一种制备a-晶型丙炔氟草胺的溶有丙炔氟草胺的热溶液的温度,以及制备a-晶型和2nd-晶型混合的丙炔氟草胺的溶有丙炔氟草胺的热溶液的温度。
所述单烷基苯与上文描述一致,在此不再赘述。
本发明将处于第二温度的溶有丙炔氟草胺的热溶液降温至结晶温度进行结晶,第二温度只要能够确保丙炔氟草胺在进行结晶前不会析出即可,一般地,所述第二温度为80-120℃,优选情况下,所述热溶液的溶剂为ch3cooh,所述第二温度为90-110℃;所述热溶液的溶剂为单烷基苯,所述第二温度为90-110℃。
根据本发明的方法,优选情况下,所述结晶温度为50-60℃,在该温度下结晶速度较快,且获得的a-晶型和和2nd-晶型混合的丙炔氟草胺的收率和纯度较高。
根据本发明的方法,结晶的时间可以根据结晶温度进行合理的选择,一般地,所述结晶的持续时间可以为1-3小时。进一步优选地,所述结晶在伴随搅拌的状态下进行。
根据本发明的方法,所述溶剂的用量以能够将丙炔氟草胺完全溶解为准,一般地,相对于1g的丙炔氟草胺,所述溶剂的用量可以为1-20g,为了获得更高收率和更高纯度的a-晶型和和2nd-晶型混合的丙炔氟草胺,优选情况下,相对于1g的丙炔氟草胺,所述溶剂的用量可以为3-11g。
本发明的方法还包括将结晶之后所得混合物降温至环境温度进行固液分离的步骤,以将a-晶型和和2nd-晶型混合的丙炔氟草胺晶体分离出来。所述固液分离的方法与上文描述一致,在此不再赘述。优选情况下,所述方法还包括将经固液分离得到的固体进行洗涤和干燥。其中,所述环境温度可以为10-30℃。
根据本发明的方法,所述热溶液可以通过以下方法制备:将丙炔氟草胺溶解于溶剂中,可选地,将部分溶剂去除。所述溶解的温度可以根据溶剂的种类进行合理的选择,一般地,所述溶解的温度可以为80-120℃。优选情况下,所述溶剂为ch3cooh,所述溶解的温度为90-110℃;溶解优选在加热回流的状态下进行,所述加热回流的时间可以为0.1-5小时,优选为0.5-2小时。所述溶剂为单烷基苯,所述溶解的温度为90-110℃;溶解优选在加热回流的状态下进行,所述加热回流的时间可以为0.1-5小时,优选为0.5-2小时。为了加快溶解,所述溶解优选在搅拌状态下进行。
根据本发明的方法,可以根据热溶液中的溶剂的用量,将所述热溶液在降温至结晶温度之前进行去除部分溶剂的步骤,可提高a-晶型和和2nd-晶型混合的丙炔氟草胺的收率和纯度。去除溶剂的方法可以为本领域中常用的方法,例如常压蒸馏,一般地,所述溶剂为ch3cooh,常压蒸馏的温度可以为115-130℃;所述溶剂为单烷基苯时,常压蒸馏的温度可以为108-115℃。
根据本发明的方法,所述丙炔氟草胺可以通过商购得到,也可以通过本领域中常用的合成方法进行制备。
根据本发明第四方面,本发明提供一种丙炔氟草胺的制备方法,该方法包括:在催化剂的存在下,将式(1)所示的6-氨基-7-氟-4-丙炔基-1,4-苯并恶嗪-3(4h)-酮与式(2)所示的3,4,5,6-四氢苯酐在第一溶剂中接触反应,所述催化剂为碱性含氮有机物、或者有机酸和碱性含氮有机物的混合物,其中,所述方法还包括采用上述a-晶型丙炔氟草胺的制备方法或者a-晶型和2nd-晶型混合的丙炔氟草胺的制备方法将接触反应得到的丙炔氟草胺进行纯化,
根据本发明的方法,所述有机酸以能够与碱性含氮有机物配合使用催化式(1)所示的6-氨基-7-氟-4-丙炔基-1,4-苯并恶嗪-3(4h)-酮与式(2)所示的3,4,5,6-四氢苯酐反应生成丙炔氟草胺为准。优选情况下,所述有机酸为ch3cooh、
根据本发明的方法,所述碱性含氮有机物以能够起到催化的6-氨基-7-氟-4-丙炔基-1,4-苯并恶嗪-3(4h)-酮与3,4,5,6-四氢苯酐反应生成丙炔氟草胺为准,一般地,所述碱性含氮有机物为选自具有式(3)、式(4)和式(5)所示结构的碱性含氮有机化合物中的一种或多种,
式(3)中,r1、r2和r3各自独立为c1-c5的烷基,优选为c2-c3的烷基;式(4)中,r4为h或c1-c5的烷基,优选为h或甲基;式(5)中,r5为h、c1-c5的烷基或-nr6r7基团,优选为-nr6r7基团,r6和r7各自独立为h或c1-c3的烷基;更优选地,所述碱性含氮有机物为选自三乙胺、哌啶和4-二甲氨基吡啶中的一种或多种。
在本发明中,进一步优选地,所述催化剂为有机酸和碱性含氮有机物的混合物,例如乙酸与三乙胺的混合物、乙酸与哌啶的混合物,以及乙酸与4-二甲氨基吡啶的混合物,优选为乙酸与哌啶的混合物。所述有机酸和碱性含氮有机物的重量比例如可以为1:0.01-5,优选为1:0.4-0.8。
根据本发明的方法,所述催化剂的用量以能够催化6-氨基-7-氟-4-丙炔基-1,4-苯并恶嗪-3(4h)-酮与3,4,5,6-四氢苯酐反应生成丙炔氟草胺为准,一般地,相对于100重量份的所述6-氨基-7-氟-4-丙炔基-1,4-苯并恶嗪-3(4h)-酮,所述催化剂的用量为0.1-100重量份,优选为1-15重量份。
根据本发明的方法,所述6-氨基-7-氟-4-丙炔基-1,4-苯并恶嗪-3(4h)-酮和3,4,5,6-四氢苯酐的用量以能够发生反应生成丙炔氟草胺为准,一般地,6-氨基-7-氟-4-丙炔基-1,4-苯并恶嗪-3(4h)-酮和3,4,5,6-四氢苯酐的用量的摩尔比可以为1:0.95-1.5,优选为1:1-1.05。
在本发明中,所述第一溶剂可以为起到溶解分散作用的有机溶剂,为了进一步提高产品的收率和纯度,优选地,所述第一溶剂为1,2-二氯乙烷、甲苯和甲基异丁基甲酮中的一种或多种,进一步优选为1,2-二氯乙烷和/或甲苯。
在本发明中,所述第一溶剂的用量以起到反应媒介的作用为准,例如,相对于100g的所述6-氨基-7-氟-4-丙炔基-1,4-苯并恶嗪-3(4h)-酮,所述第一溶剂的用量可以为300-800ml,优选为400-600ml。
根据本发明的方法,所述接触的条件以能够生成丙炔氟草胺为准,一般地,所述接触在60-120℃下进行,优选在90-120℃下进行。所述接触的时间可以根据接触的温度进行合理的选择,一般地,所述接触的时间为1-24h,优选为3-5h。
在本发明中,从所述接触反应所得混合物中获得丙炔氟草胺产品的方式没有特别的限定,例如可以将上述接触反应后所得的混合物冷却至丙炔氟草胺析出然后过滤得到固相的丙炔氟草胺产品,根据本发明的一种优选实施方式,也可以将所述所得混合物先减压蒸馏掉一半以上的溶剂或利用与水共沸脱除溶剂,然后冷却至室温使丙炔氟草胺析出,进而过滤得到固相的丙炔氟草胺产品。
根据本发明的a-晶型丙炔氟草胺的制备方法,所述丙炔氟草胺的制备方法还包括采用上述方法对接触反应得到的丙炔氟草胺进行纯化,得到a-晶型丙炔氟草胺。
根据本发明的a-晶型和2nd-晶型混合的丙炔氟草胺的制备方法,所述丙炔氟草胺的方法还包括采用上述方法对接触反应得到的丙炔氟草胺进行纯化,得到a-晶型和2nd-晶型混合的丙炔氟草胺。
根据本发明的一种优选实施方式,当第一有机溶剂为甲苯时,可以直接将接触反应得到的混合物可选地分离出部分溶剂后,采用本发明的上述方法进行结晶。
在一种实施方式中,可以在甲苯的存在下,在催化剂的存在下,将式(1)所示的6-氨基-7-氟-4-丙炔基-1,4-苯并恶嗪-3(4h)-酮与式(2)所示的3,4,5,6-四氢苯酐在60-120℃下(优选为90-120℃)接触反应,反应1-24小时(优选3-5小时)后,降温至80-100℃下(优选为80-90℃)结晶,结晶的持续时间为1-3小时,然后将结晶之后所得的混合物降温至环境温度进行固液分离得到a-晶型丙炔氟草胺,其中,所述催化剂为碱性含氮有机物或者有机酸和碱性含氮有机物的混合物。
在另一种实施方式中,可以在甲苯的存在下,在催化剂的存在下,将式(1)所示的6-氨基-7-氟-4-丙炔基-1,4-苯并恶嗪-3(4h)-酮与式(2)所示的3,4,5,6-四氢苯酐在60-120℃下(优选为90-120℃)接触反应,反应1-24小时(优选3-5小时)后,降温至50-65℃(优选为50-60℃)下结晶,结晶的持续时间为1-3小时,然后将结晶之后所得的混合物降温至环境温度进行固液分离得到a-晶型和2nd-晶型混合的丙炔氟草胺。
根据本发明的第五方面,本发明还提供了一种丙炔氟草胺的制备方法,该方法包括:将式(1)所示的6-氨基-7-氟-4-丙炔基-1,4-苯并恶嗪-3(4h)-酮与式(2)所示的3,4,5,6-四氢苯酐在第二溶剂中接触反应,所述第二溶剂为ch3cooh,其中,所述方法还包括采用上述a-晶型丙炔氟草胺的制备方法或者a-晶型和2nd-晶型混合的丙炔氟草胺的制备方法对接触反应得到的丙炔氟草胺进行纯化。
根据本发明的方法,ch3cooh的提供方式与上文描述相同,在此不再赘述。
根据本发明的方法,所述第二溶剂的用量以起到反应媒介的作用为准,例如,相对于100g的所述6-氨基-7-氟-4-丙炔基-1,4-苯并恶嗪-3(4h)-酮,所述第二溶剂的用量可以为300-800ml,优选为300-600ml。
所述6-氨基-7-氟-4-丙炔基-1,4-苯并恶嗪-3(4h)-酮和3,4,5,6-四氢苯酐的用量与上文描述相同,在此不再赘述。
根据本发明的方法,所述接触的条件以能够生产丙炔氟草胺为准,一般地,所述接触可以在80-120℃下进行,优选在90-120℃下进行。所述接触的时间可以根据接触的温度进行合理的选择,一般地,所述接触的时间为2-6h,优选为3-5h。所述接触优选在加热回流的状态下进行。
根据本发明的a-晶型丙炔氟草胺的制备方法,所述丙炔氟草胺的制备方法还包括采用上述方法将接触反应得到的丙炔氟草胺进行纯化,得到a-晶型丙炔氟草胺。
根据本发明的a-晶型和2nd-晶型混合的丙炔氟草胺的制备方法,所述丙炔氟草胺的方法还包括采用上述方法将接触反应得到的丙炔氟草胺进行纯化,得到a-晶型和2nd-晶型混合的丙炔氟草胺。
根据本发明的一种优选实施方式,当第二溶剂为ch3cooh时,可以直接将接触反应得到的混合物可选地分离出部分溶剂后,采用本发明所述的方法进行结晶。
在一种实施方式中,可以在ch3cooh的存在下,将式(1)所示的6-氨基-7-氟-4-丙炔基-1,4-苯并恶嗪-3(4h)-酮与式(2)所示的3,4,5,6-四氢苯酐在80-120℃下(优选为90-120℃)接触反应,反应2-6小时(优选为3-5小时)后,降温至80-100℃(优选为80-90℃)下结晶,结晶的持续时间为1-3小时,然后将结晶之后所得的混合物降温至环境温度进行固液分离得到a-晶型丙炔氟草胺。
在另一种实施方式中,可以在ch3cooh的存在下,将式(1)所示的6-氨基-7-氟-4-丙炔基-1,4-苯并恶嗪-3(4h)-酮与式(2)所示的3,4,5,6-四氢苯酐在80-120℃下(优选为90-120℃)接触反应,反应2-6小时(优选为3-5小时)后,降温至50-65℃(优选为50-60℃)下结晶,结晶的持续时间为1-3小时,然后将结晶之后所得的混合物降温至环境温度进行固液分离得到a-晶型和2nd-晶型混合的丙炔氟草胺。
以下将通过实施例对本发明进行详细描述。
(1)以下实施例和对比例中,粉末x射线衍射的测试条件包括cukα辐射,石墨单色器,管压为40kv,管流为150ma,发散狭缝ds为1°,散射狭缝ss为1°,接收狭缝rs为0.15mm,扫描范围为5°-30°,扫描速度为3°·min-1,步长0.02°。
(2)以下实施例和对比例中,dsc测定的条件包括:铝坩埚,al2o3作参比物,测试温度为25-350℃,升温速率为5℃/min。样品用量为约5mg。
(3)以下实施例和对比例中,x倍当量指的是相对于1摩尔的6-氨基-7-氟-4-丙炔基-1,4-苯并恶嗪-3(4h)-酮,其用量为x摩尔。
在实施例中,在进行反应之前,先对原料6-氨基-7-氟-4-丙炔基-1,4-苯并恶嗪-3(4h)-酮和3,4,5,6-四氢苯酐进行纯化的预处理,使其含量能够达到99%左右,具体的方法如下;
3,4,5,6-四氢苯酐的纯化方法参照欧洲专利ep0432797b1所公开的方法;
6-氨基-7-氟-4-丙炔基-1,4-苯并恶嗪-3(4h)-酮的纯化方法包括:取41.6g粗品加入反应瓶,加800ml水,慢慢滴加盐酸76ml全溶ph=1,加活性炭4.5g,升温到60℃搅拌15分钟,过滤(机械杂质),滤液加1.5倍量的乙酸乙酯萃取分层,水相用氨水调ph到中性(有固体吸出),过滤,滤饼再用230ml乙醇升温回流重结晶,自然降温(针状晶体),得干品16g,定量:99%;针对实际情况所需的量按照上述用量比换算。
实施例1-12用于说明本发明提供的a-晶型丙炔氟草胺的制备方法。
实施例1
取10g的纯度为99重量%的6-氨基-7-氟-4-丙炔基-1,4-苯并恶嗪-3(4h)-酮以及30ml醋酸(浓度为99重量%,以下相同)、1.05倍当量的纯度为99.5重量%的3,4,5,6-四氢苯酐置于反应瓶中,95℃下反应3.5小时,降温至80℃,保温缓慢结晶,搅拌2小时,降温至20℃过滤,将得到的固体进行洗涤和干燥得到a-晶型丙炔氟草胺。
将得到的a-晶型丙炔氟草胺进行x射线衍射谱图和dsc测定,其中,图1为该a-晶型丙炔氟草胺的dsc图,图2为该a-晶型丙炔氟草胺的x射线衍射谱图。该x射线衍射谱图的衍生数据与cn104169274a中的表2的数据一致,由图1和图2表明得到a-晶型丙炔氟草胺。
实施例2
取10g的纯度为99重量%的6-氨基-7-氟-4-丙炔基-1,4-苯并恶嗪-3(4h)-酮以及30ml醋酸、1.05倍当量的纯度为99.5重量%的3,4,5,6-四氢苯酐置于反应瓶中,100℃下反应4小时,降温至85℃,保温缓慢结晶,搅拌3小时,降温至25℃过滤,将得到的固体进行洗涤和干燥得到a-晶型丙炔氟草胺。
将得到的a-晶型丙炔氟草胺进行x射线衍射谱图和dsc测定,其x射线衍射谱图和dsc图与实施例1相同,表明得到a-晶型丙炔氟草胺。
实施例3
取10g的纯度为98重量%的丙炔氟草胺和30ml醋酸,置于反应瓶中,加热至110℃下回流0.5小时,降温至90℃,保温缓慢结晶,搅拌2小时,降温至23℃过滤,将得到的固体进行洗涤和干燥得到a-晶型丙炔氟草胺。
将得到的a-晶型丙炔氟草胺进行x射线衍射谱图和dsc测定,其x射线衍射谱图和dsc图与实施例1相同,表明得到a-晶型丙炔氟草胺。
实施例4
取10g的纯度为98重量%的丙炔氟草胺和100ml甲苯,置于反应瓶中,加热至110℃下回流1小时,降温至80℃,保温缓慢结晶,搅拌2小时,降温至23℃过滤,将得到的固体进行洗涤和干燥得到a-晶型的丙炔氟草胺。
将得到的a-晶型丙炔氟草胺进行x射线衍射谱图和dsc测定,其x射线衍射谱图和dsc图与实施例1相同,表明得到a-晶型丙炔氟草胺。
实施例5
取1g的纯度为98重量%的丙炔氟草胺和10ml甲苯,置于反应瓶中,加热至98℃下保温2小时,降温至85℃,保温缓慢结晶,搅拌3小时,降温至23℃过滤,将得到的固体进行洗涤和干燥得到a-晶型的丙炔氟草胺。
将得到的a-晶型丙炔氟草胺进行x射线衍射谱图和dsc测定,其x射线衍射谱图和dsc图与实施例1相同,表明得到a-晶型丙炔氟草胺。
实施例6
采用与实施例4相同的方法,不同的是,加热至110℃下回流1小时,常压蒸馏去除甲苯至所得混合物至25ml后,降温至80℃,保温缓慢结晶,搅拌2小时,降温至23℃过滤,将得到的固体进行洗涤和干燥得到a-晶型的丙炔氟草胺。
将得到的a-晶型丙炔氟草胺进行x射线衍射谱图和dsc测定,其x射线衍射谱图和dsc图与实施例1相同,表明得到a-晶型丙炔氟草胺。
实施例7
取4.5g的纯度为98重量%的丙炔氟草胺和50g丙酮,置于反应瓶中,加热至62℃下回流0.5小时至全溶后,在加热状态下缓慢加入100ml的25℃的水进行结晶,结晶的持续时间为2.5小时,在62℃下常压蒸馏除去丙酮,降温至23℃过滤,将得到的固体进行洗涤和干燥得到a-晶型丙炔氟草胺。
将得到的a-晶型丙炔氟草胺进行x射线衍射谱图和dsc测定,其x射线衍射谱图和dsc图与实施例1相同,表明得到a-晶型丙炔氟草胺。
实施例8
取4.5g的纯度为98重量%的丙炔氟草胺和66.2g四氢呋喃,置于反应瓶中,加热至70℃下回流1小时至全溶后,在加热状态下缓慢加入88.2ml的25℃的水进行结晶,结晶的持续时间为1小时,在70℃下常压蒸馏除去四氢呋喃,降温至25℃过滤,将得到的固体进行洗涤和干燥得到a-晶型丙炔氟草胺。
将得到的a-晶型丙炔氟草胺进行x射线衍射谱图和dsc测定,其x射线衍射谱图和dsc图与实施例1相同,表明得到a-晶型丙炔氟草胺。
实施例9
取4.5g的98%含量丙炔氟草胺和30g的n,n-二甲基甲酰胺,置于反应瓶中,加热至80℃保温2小时至全溶后,在加热状态下缓慢加入110ml的25℃水进行结晶,结晶的持续时间为1.5小时,降温至20℃过滤,得到a-晶型丙炔氟草胺。
将得到的a-晶型丙炔氟草胺进行x射线衍射谱图和dsc测定,其x射线衍射谱图和dsc图与实施例1相同,表明得到a-晶型丙炔氟草胺。
实施例10
取20g纯度为99重量%的6-氨基-7-氟-4-丙炔基-1,4-苯并恶嗪-3(4h)-酮、100ml1,2-二氯乙烷、1.05倍当量的纯度为99.5重量%的3,4,5,6-四氢苯酐、1.4ml乙酸和1ml哌啶置于反应瓶中,100℃下保温反应7小时共沸脱水,反应完毕,加入水共沸脱出1,2-二氯乙烷,过滤,将所得滤饼用95%乙醇打浆,过滤,干燥得丙炔氟草胺,纯度为99.5重量%。将制得的丙炔氟草胺进行dsc和x射线衍射谱图测定,结果表明,该丙炔氟草胺为a-晶型和2nd晶型混合物。
取10g的上述丙炔氟草胺和30ml醋酸,置于反应瓶中,加热至110℃下保温0.5小时,降温至90℃,保温缓慢结晶,搅拌2小时,降温至23℃过滤,将得到的固体进行洗涤和干澡得到a-晶型丙炔氟草胺。
将得到的a-晶型丙炔氟草胺进行x射线衍射谱图和dsc测定,其x射线衍射谱图和dsc图与实施例1相同,表明得到a-晶型丙炔氟草胺。
实施例11
取20g纯度为99重量%的6-氨基-7-氟-4-丙炔基-1,4-苯并恶嗪-3(4h)-酮以及80ml甲苯、1.1倍当量的纯度为99.5重量%的3,4,5,6-四氢苯酐、1.4ml乙酸和1.2ml哌啶置于反应瓶中,110℃下回流反应6小时共沸脱水,反应完毕,加入水共沸脱出甲苯,过滤,将所得滤饼用95%乙醇打浆,过滤,干燥得到丙炔氟草胺,纯度为99.3重量%。将制得的丙炔氟草胺进行dsc和x射线衍射谱图测定,结果表明,该丙炔氟草胺为a-晶型和2nd晶型混合物。
取10g的上述丙炔氟草胺和100ml甲苯,置于反应瓶中,加热至110℃下回流0.5小时,降温至90℃,保温缓慢结晶,搅拌3小时,降温至23℃下过滤,将得到的固体进行洗涤和干澡得到a-晶型丙炔氟草胺。
将得到的a-晶型丙炔氟草胺进行x射线衍射谱图和dsc测定,其x射线衍射谱图和dsc图与实施例1相同,表明得到a-晶型丙炔氟草胺。
实施例12
取10g的实施例11中制备的纯度为99.3重量%的丙炔氟草胺和100g丙酮,置于反应瓶中,加热至65℃下回流0.5小时至全溶后,在加热状态下缓慢加入100ml的25℃的水进行结晶,结晶的持续时间为2小时,在65℃下常压蒸馏除去丙酮,降温至23℃过滤,将得到的固体进行洗涤和干燥得到a-晶型丙炔氟草胺。
将得到的a-晶型丙炔氟草胺进行x射线衍射谱图和dsc测定,其x射线衍射谱图和dsc图与实施例1相同,表明得到a-晶型丙炔氟草胺。
实施例13-17用于说明本发明提供的a-晶型和2nd-晶型混合的丙炔氟草胺的制备方法。
实施例13
取10g的纯度为98重量%的丙炔氟草胺和30ml醋酸,置于反应瓶中,加热至95℃保温0.5小时,降温至50℃,保温缓慢结晶,搅拌3小时,降温至23℃过滤,将得到的固体进行洗涤和干燥得到a-晶型和2nd-晶型混合的丙炔氟草胺。
将得到的a-晶型和2nd-晶型混合的丙炔氟草胺进行x射线衍射谱图和dsc测定,其中,图3为该a-晶型和2nd-晶型混合的丙炔氟草胺的dsc图,图4为该a-晶型和2nd-晶型混合的丙炔氟草胺的x射线衍射谱图,表明得到a-晶型和2nd-晶型混合的丙炔氟草胺。
实施例14
取10g纯度为98重量%的丙炔氟草胺和100ml甲苯,置于反应瓶中,加热至100℃回流1小时,降温至55℃,保温缓慢结晶,搅拌2小时,降温至23℃过滤,将得到的固体进行洗涤和干燥得到a-晶型和2nd-晶型混合的丙炔氟草胺。
将得到的a-晶型和2nd-晶型混合的丙炔氟草胺进行x射线衍射谱图和dsc测定,其x射线衍射谱图和dsc图与实施例13相同,表明得到a-晶型和2nd-晶型混合的丙炔氟草胺。
实施例15
取10g纯度为99重量%的6-氨基-7-氟-4-丙炔基-1,4-苯并恶嗪-3(4h)-酮以及30ml醋酸、1.05倍当量的纯度为99.5重量%的3,4,5,6-四氢苯酐置于反应瓶中,99℃下反应3.5小时,降温至60℃,保温缓慢结晶,搅拌1小时,降温至20℃过滤,将得到的固体进行洗涤和干燥得到a-晶型和2nd-晶型混合的丙炔氟草胺。
将得到的a-晶型和2nd-晶型混合的丙炔氟草胺进行x射线衍射谱图和dsc测定,其x射线衍射谱图和dsc图与实施例13相同,表明得到a-晶型和2nd-晶型混合的丙炔氟草胺。
实施例16
取10g实施例10中制备的纯度为99.5重量%的丙炔氟草胺和30ml醋酸,置于反应瓶中,加热至110℃保温0.5小时,降温至55℃,保温缓慢结晶,搅拌2小时,降温至23℃过滤,将得到的固体进行洗涤和干燥得到a-晶型和2nd-晶型混合的丙炔氟草胺。
将得到的a-晶型和2nd-晶型混合的丙炔氟草胺进行x射线衍射谱图和dsc测定,其x射线衍射谱图和dsc图与实施例13相同,表明得到a-晶型和2nd-晶型混合的丙炔氟草胺。
实施例17
取10g实施例11中制备的纯度为99.3重量%的丙炔氟草胺和100ml甲苯,置于反应瓶中,加热至110℃回流1小时,降温至50℃,保温缓慢结晶,搅拌2小时,降温至23℃过滤,将得到的固体进行洗涤和干燥得到a-晶型和2nd-晶型混合的丙炔氟草胺。
将得到的a-晶型和2nd-晶型混合的丙炔氟草胺进行x射线衍射谱图和dsc测定,其x射线衍射谱图和dsc图与实施例13相同,表明得到a-晶型和2nd-晶型混合的丙炔氟草胺。
对比例1
取5g纯度为98重量%的丙炔氟草胺和50ml二氯甲烷,置于反应瓶中,23℃下搅拌至全溶,然后在23℃下滴加500ml正己烷,保温缓慢结晶1小时,然后过滤,将得到的固体进行洗涤和干燥得到a-晶型和2nd-晶型混合的丙炔氟草胺。
对比例2
采用对比例1的方法,不同的是,在-20℃下滴加500ml正己烷。
将得到的a-晶型和2nd-晶型混合的丙炔氟草胺进行x射线衍射谱图和dsc测定,确定该固体为a-晶型和2nd-晶型混合的丙炔氟草胺。
对比例3
根据us4640707中实施例2的方法制备丙炔氟草胺,并按该专利中记载柱层析方法得到固体,经x射线衍射和dsc测定确定该固体为a-晶型和2nd-晶型混合的丙炔氟草胺。
以上详细描述了本发明的优选实施方式,但是,本发明并不限于上述实施方式中的具体细节,在本发明的技术构思范围内,可以对本发明的技术方案进行多种简单变型,这些简单变型均属于本发明的保护范围。
另外需要说明的是,在上述具体实施方式中所描述的各个具体技术特征,在不矛盾的情况下,可以通过任何合适的方式进行组合。为了避免不必要的重复,本发明对各种可能的组合方式不再另行说明。
此外,本发明的各种不同的实施方式之间也可以进行任意组合,只要其不违背本发明的思想,其同样应当视为本发明所公开的内容。
- 还没有人留言评论。精彩留言会获得点赞!