基于吩噻嗪衍生物的室温磷光分子及其制备方法和应用与流程
本发明属于有机化学领域,具体涉及一类基于吩噻嗪衍生物的室温磷光分子及其制备方法和作为防伪材料的应用。
背景技术:
磷光材料在防伪、生物成像、分子传感、光催化反应和光伏器件等诸多高新科技领域有着广阔的应用前景。传统室温磷光材料大多是无机化合物和有机金属,但无机化合物和有机金属存在诸多缺点,如品种有限、材料成本高、容易受损、不合适大面积制备、稳定性差等。而纯有机分子磷光材料相对便宜、结构容易调节、对环境相对安全且具有良好的加工性,这使得它们更适用于生物工程、医药技术领域、传感器和电子器件的制备。但是目前报道的纯有机磷光材料种类非常少,特别是长寿命的纯有机室温磷光材料更是十分稀缺,因此,如何开发长寿命的纯有机室温磷光材料成为此领域研究的重点和难点。
技术实现要素:
本发明的目的在于发展一类长寿命的有机室温磷光分子,提供一种基于吩噻嗪衍生物的室温磷光分子及其制备方法和作为防伪材料的应用。
本发明提供的技术方案具体如下:
一种基于吩噻嗪衍生物的室温磷光分子,具有式(i)所示的结构:
其中,r为-och3、-ch3、-h、-br、-cl、-f或-cf3。
一种制备基于吩噻嗪衍生物的室温磷光分子的方法,包括以下步骤:
(1)氮气保护下,将吩噻嗪、化合物a、叔丁醇钾、催化量的醋酸钯、催化量的三叔丁基膦溶于足量的甲苯溶液中,在110-120℃下搅拌回流12-24h,反应完毕后,将反应液冷却至室温,纯化,得到中间产物b,即
所述的化合物a为4-甲氧基碘苯、4-甲基碘苯、碘苯、4-溴碘苯、4-氯碘苯、4-氟碘苯、4-三氟甲基碘苯中的一种;
(2)将中间产物b用二氯甲烷、醋酸和双氧水的混合液溶解,在70℃下反应12-24h,纯化,得到式(i)所示结构的化合物。
所述吩噻嗪、化合物a、叔丁醇钾的摩尔比为10:12:15。
所述醋酸钯和三叔丁基膦的摩尔比为2:1。
所述二氯甲烷、醋酸和双氧水的混合液中二氯甲烷、醋酸和双氧水的体积比为90:45:2。
步骤(1)中的纯化方式为:将反应液冷却至室温,然后用三氯甲烷萃取,收集有机相,再用无水na2so4干燥,旋干,得到粗产品;以石油醚为淋洗剂,将粗产品用硅胶色谱柱层析进行分离纯化,即成。
步骤(2)中的纯化方式为:反应液用三氯甲烷萃取,收集有机相,再用无水na2so4干燥,旋干,得到粗产品;以体积比为1:1的石油醚和二氯甲烷的混合液为淋洗剂,将粗产品用硅胶色谱柱层析分离,即成。
上述基于吩噻嗪衍生物的室温磷光分子在防伪领域中的应用。
本发明主要是以吩噻嗪为核通过碳氮偶联反应将苯环衍生物与之相连,再将其氧化,得到目标产物。该制备方法的反应条件温和,产率较高。而且它们的固体粉末均可发生室温磷光现象,磷光寿命在室温条件下最高长达398ms。由于分子的这种室温磷光特性,它们可以作为防伪材料使用。
本发明具有以下优点和有益效果:
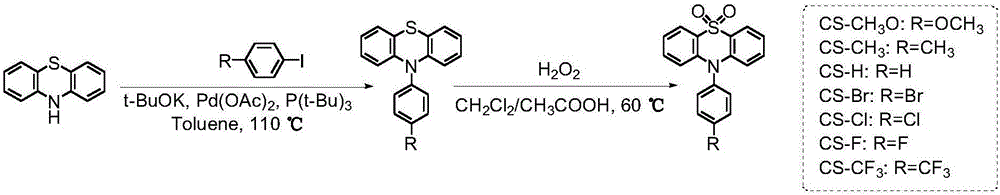
(1)本发明的化合物制备方法的反应条件温和,产率较高,如图1所示。
(2)该类化合物的固体粉末均有室温磷光现象,如图2所示,且它们的最长寿命达398ms,
可作为防伪材料。
附图说明
图1为化合物cs-ch3o、cs-ch3、cs-h、cs-br、cs-cl、cs-f和cs-cf3的合成路线图。
图2为化合物cs-ch3o、cs-ch3、cs-h、cs-br、cs-cl、cs-f和cs-cf3的室温磷光发光图。
图3为化合物cs-ch3o、cs-ch3、cs-h、cs-br、cs-cl、cs-f和cs-cf3的室温磷光光谱以及磷光寿命图;其中,图3(a)为磷光光谱图,图3(b)为磷光寿命图。
图4为cs-f和4,4'-二溴二苯甲酮组成的图案在紫外灯照射前后的对比图。
具体实施方式
下面结合具体实施例对本发明做进一步详细的描述。本领域技术人员应理解,下面的实施例仅用于本说明的技术方案,并不用于限制本发明的保护范围。
下述实施例中所用的原料为市售产品,或可用本领域已知的方法合成得到。
实施例1:化合物cs-ch3o的合成
在氮气保护下,在反应瓶中,将吩噻嗪(1.99g,10mmol)、4-甲氧基碘苯(2.34g,12mol)、叔丁醇钾(1.68g,15mmol)和醋酸钯(0.11g,0.5mmol)、三叔丁基膦(0.5ml,0.25mmol)溶于足量的甲苯溶液中,在110-120℃下搅拌回流12h,反应完毕后,将反应液冷却至室温,然后用三氯甲烷萃取,收集有机相,再用无水na2so4干燥,旋干,得到粗产品。以石油醚为淋洗剂,将粗产品用硅胶色谱柱层析进行分离纯化,再将得到的产物用二氯甲烷(90ml)、醋酸(45ml)和双氧水(2ml)的混合液溶解,在70℃下反应12h,反应液用三氯甲烷萃取,收集有机相,再用无水na2so4干燥,旋干,得到粗产品。以石油醚和二氯甲烷的混合液(v/v,1/1)为淋洗剂,将粗产品用硅胶色谱柱层析进行分离纯化,得白色固体(2.69g,80%),并用1hnmr、13cnmr和ms对结构进行表征,证实该白色固体为化合物cs-ch3o。
1h-nmr(300mhz,cdcl3)δ(ppm):8.15-8.17(d,2h),7.36-7.39(t,2h),7.16-7.30(m,6h),6.66-6.69(d,2h),3.94(s,3h).13c-nmr(75mhz,cdcl3)δ(ppm):160.4,141.3,133.0,131.6,131.4,123.5,122.7,122.2,117.6,116.6,55.9.ms(ei),m/z:336.98([m+],calcdforc19h15no3s,337.08。
实施例2:化合物cs-ch3的合成
在氮气保护下,在反应瓶中,将吩噻嗪(1.99g,10mmol)、4-甲基碘苯(2.62g,12mol)、叔丁醇钾(1.68g,15mmol)和醋酸钯(0.11g,0.5mmol)、三叔丁基膦(0.5ml,0.25mmol)溶于足量的甲苯溶液中,在110-120℃下搅拌回流15h,反应完毕后,将反应液冷却至室温,然后用三氯甲烷萃取,收集有机相,再用无水na2so4干燥,旋干,得到粗产品。以石油醚为淋洗剂,将粗产品用硅胶色谱柱层析进行分离纯化,再将得到的产物用二氯甲烷(90ml)、醋酸(45ml)和双氧水(2ml)的混合液溶解,在70℃下反应16h,反应液用三氯甲烷萃取,收集有机相,再用无水na2so4干燥,旋干,得到粗产品。以石油醚和二氯甲烷的混合液(v/v,1/1)为淋洗剂,将粗产品用硅胶色谱柱层析进行分离纯化,得白色固体(2.31g,72%),并用1hnmr、13cnmr和ms对结构进行表征,证实该白色固体为化合物cs-ch3。
1h-nmr(300mhz,cdcl3)δ(ppm):8.15-8.18(dd,2h),7.47-7.50(d,2h),7.35-7.41(m,2h),7.21-7.26(m,4h),6.64-6.67(d,2h),2.52(s,3h).13c-nmr(75mhz,cdcl3)δ(ppm):141.1,140.1,136.4,132.9,132.2,130.3,123.5,122.7,122.1,117.5,21.6.ms(ei),m/z:320.98([m+],理论值:c19h15no2s,321.08。
实施例3:化合物cs-h的合成
在氮气保护下,在反应瓶中,将吩噻嗪(1.99g,10mmol)、碘苯(2.45g,12mol)、叔丁醇钾(1.68g,15mmol)和醋酸钯(0.11g,0.5mmol)、三叔丁基膦(0.5ml,0.25mmol)溶于足量的甲苯溶液中,在110-120℃下搅拌回流20h,反应完毕后,将反应液冷却至室温,然后用三氯甲烷萃取,收集有机相,再用无水na2so4干燥,旋干,得到粗产品。以石油醚为淋洗剂,将粗产品用硅胶色谱柱层析进行分离纯化,再将得到的产物用二氯甲烷(90ml)、醋酸(45ml)和双氧水(2ml)的混合液溶解,在70℃下反应18h,反应液用三氯甲烷萃取,收集有机相,再用无水na2so4干燥,旋干,得到粗产品。以石油醚和二氯甲烷的混合液(v/v,1/1)为淋洗剂,将粗产品用硅胶色谱柱层析进行分离纯化,得白色固体(2.61g,85%),并用1hnmr、13cnmr和ms对结构进行表征,证实该白色固体为化合物cs-h。
1h-nmr(300mhz,cdcl3)δ(ppm):8.16-8.19(d,2h),7.63-7.73(m,3h),7.36-7.40(m,4h),7.22-7.26(m,2h),6.61-6.63(d,2h).13c-nmr(75mhz,cdcl3)δ(ppm):140.7,138.8,132.7,131.2,130.3,129.7,123.3,122.5,121.9,117.1.ms(ei),m/z:306.97([m+],理论值:c18h13no2s,307.07。
实施例4:化合物cs-br的合成
在氮气保护下,在反应瓶中,将吩噻嗪(1.99g,10mmol)、4-溴碘苯(3.38g,12mol)、叔丁醇钾(1.68g,15mmol)和醋酸钯(0.11g,0.5mmol)、三叔丁基膦(0.5ml,0.25mmol)溶于足量的甲苯溶液中,在110-120℃下搅拌回流24h,反应完毕后,将反应液冷却至室温,然后用三氯甲烷萃取,收集有机相,再用无水na2so4干燥,旋干,得到粗产品。以石油醚为淋洗剂,将粗产品用硅胶色谱柱层析进行分离纯化,再将得到的产物用二氯甲烷(90ml)、醋酸(45ml)和双氧水(2ml)的混合液溶解,在70℃下反应20h,反应液用三氯甲烷萃取,收集有机相,再用无水na2so4干燥,旋干,得到粗产品。以石油醚和二氯甲烷的混合液(v/v,1/1)为淋洗剂,将粗产品用硅胶色谱柱层析进行分离纯化,得白色固体(2.12g,55%),并用1hnmr、13cnmr和ms对结构进行表征,证实该白色固体为化合物cs-br。
1h-nmr(400mhz,cdcl3)δ(ppm):8.16-8.19(m,2h),7.83-7.85(m,2h),7.39-7.43(m,2h),7.25-7.30(m,4h),6.61-6.64(m,2h).13c-nmr(75mhz,cdcl3)δ(ppm):140.4,137.8,134.6,132.8,132.2,123.8,123.4,122.8,122.2,116.9.ms(ei),m/z:384.83([m+],理论值:c18h12brno2s,384.98。
实施例5:化合物cs-cl的合成
在氮气保护下,在反应瓶中,将吩噻嗪(1.99g,10mmol)、4-氯碘苯(2.86g,12mol)、叔丁醇钾(1.68g,15mmol)和醋酸钯(0.11g,0.5mmol)、三叔丁基膦(0.5ml,0.25mmol)溶于足量的甲苯溶液中,在110-120℃下搅拌回流12h,反应完毕后,将反应液冷却至室温,然后用三氯甲烷萃取,收集有机相,再用无水na2so4干燥,旋干,得到粗产品。以石油醚为淋洗剂,将粗产品用硅胶色谱柱层析进行分离纯化,再将得到的产物用二氯甲烷(90ml)、醋酸(45ml)和双氧水(2ml)的混合液溶解,在70℃下反应24h,反应液用三氯甲烷萃取,收集有机相,再用无水na2so4干燥,旋干,得到粗产品。以石油醚和二氯甲烷的混合液(v/v,1/1)为淋洗剂,将粗产品用硅胶色谱柱层析进行分离纯化,得白色固体(1.53g,45%),并用1hnmr、13cnmr和ms对结构进行表征,证实该白色固体为化合物cs-cl。
1h-nmr(400mhz,cdcl3)δ(ppm):8.17-8.19(m,2h),7.67-7.70(m,2h),7.39-7.43(m,2h),7.34-7.36(m,2h),7.25-7.29(m,2h),6.61-6.63(m,2h).13c-nmr(100mhz,cdcl3)δ(ppm):140.6,137.3,135.9,132.9,132.0,131.7,123.5,122.8,122.3,117.0.ms(ei),m/z:340.92([m+],理论值:c18h12clno2s,341.03。
实施例6:化合物cs-cf的合成
在氮气保护下,在反应瓶中,将吩噻嗪(1.99g,10mmol)、4-氟碘苯(2.66g,12mol)、叔丁醇钾(1.68g,15mmol)和醋酸钯(0.11g,0.5mmol)、三叔丁基膦(0.5ml,0.25mmol)溶于足量的甲苯溶液中,在110-120℃下搅拌回流12h,反应完毕后,将反应液冷却至室温,然后用三氯甲烷萃取,收集有机相,再用无水na2so4干燥,旋干,得到粗产品。以石油醚为淋洗剂,将粗产品用硅胶色谱柱层析进行分离纯化,再将得到的产物用二氯甲烷(90ml)、醋酸(45ml)和双氧水(2ml)的混合液溶解,在70℃下反应12h,反应液用三氯甲烷萃取,收集有机相,再用无水na2so4干燥,旋干,得到粗产品。以石油醚和二氯甲烷的混合液(v/v,1/1)为淋洗剂,将粗产品用硅胶色谱柱层析进行分离纯化,得白色固体(2.18g,67%),并用1hnmr、13cnmr和ms对结构进行表征,证实该白色固体为化合物cs-f。
1h-nmr(400mhz,cdcl3)δ(ppm):8.16-8.18(m,2h),7.38-7.43(m,6h),7.24-7.28(m,2h),6.61-6.63(m,2h).13c-nmr(100mhz,cdcl3)δ(ppm):164.0,161.5,140.7,134.6,132.8,132.4,132.3,123.4,122.7,122.2,118.5,118.3,117.0.ms(ei),m/z:324.95([m+],理论值:c18h12fno2s,325.06。
实施例7:化合物cs-cf3的合成
在氮气保护下,在反应瓶中,将吩噻嗪(1.99g,10mmol)、4-三氟甲基碘苯(3.26g,12mol)、叔丁醇钾(1.68g,15mmol)和醋酸钯(0.11g,0.5mmol)、三叔丁基膦(0.5ml,0.25mmol)溶于足量的甲苯溶液中,在110-120℃下搅拌回流12h,反应完毕后,将反应液冷却至室温,然后用三氯甲烷萃取,收集有机相,再用无水na2so4干燥,旋干,得到粗产品。以石油醚为淋洗剂,将粗产品用硅胶色谱柱层析进行分离纯化,再将得到的产物用二氯甲烷(90ml)、醋酸(45ml)和双氧水(2ml)的混合液溶解,在70℃下反应12h,反应液用三氯甲烷萃取,收集有机相,再用无水na2so4干燥,旋干,得到粗产品。以石油醚和二氯甲烷的混合液(v/v,1/1)为淋洗剂,将粗产品用硅胶色谱柱层析进行分离纯化,得白色固体(2.70g,72%),并用1hnmr、13cnmr和ms对结构进行表征,证实该白色固体为化合物cs-cf3。
1h-nmr(400mhz,cdcl3)δ(ppm):8.00-8.03(t,4h),7.58-7.61(d,2h),7.40-7.45(t,2h),7.24-7.29(t,2h),6.62-6.65(d,2h).13c-nmr(100mhz,cdcl3)δ(ppm):142.14,142.13,140.31,133.01,132.22,131.89,131.43,128.67,128.63,128.60,128.56,124.88,123.64,122.91,122.54,122.17,116.88.ms(ei),m/z:374.94([m+],理论值:c19h12f3no2s,375.05。
实施例8:性能测试
如图4所示,在黑暗条件下具有室温磷光特性的cs-f和不具有室温磷光特性的4,4'-二溴二苯甲酮在紫外灯照射下(激发波长365nm)可呈现出“8”的图案,由于4,4'-二溴二苯甲酮不具有室温磷光的特性,而cs-f具有室温磷光的特性,所以当停止紫外灯照射时,4,4'-二溴二苯甲酮不再发光,而cs-f可以持续发射磷光,呈现“7”的图案。
上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受上述实施例的限制,其他的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合、简化,均应为等效的置换方式,都包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!