苯骈氮杂烷基芳基哌嗪衍生物及在制备药物中的应用的制作方法
本发明涉及一种苯骈氮杂烷基芳基哌嗪衍生物以及在制备抗抑郁、抗神经痛等药物中的应用。
背景技术:
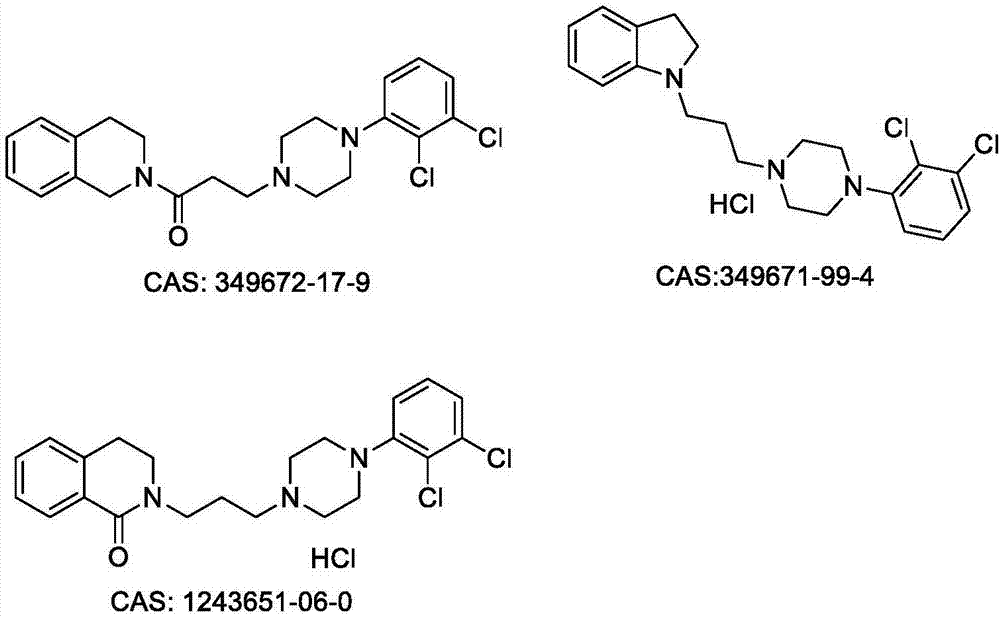
:抑郁症是一种常见的精神疾病,呈现慢性、易复发等特征。据世界卫生组织统计,全球约有3.5亿人口遭受抑郁症的折磨。预计到2020年,抑郁症将成为第二大影响人类生活质量的疾病。目前,抗抑郁症药物的作用机制尚未完全阐明。有明确疗效的药物基本作用于神经末梢突触部位,通过调节突触间隙神经递质的水平而发挥治疗作用。其病因学的生化研究表明抑郁症主要与中枢5-羟色胺(5-ht)、去甲肾上腺素(na)、多巴胺(da)、乙酰胆碱(ach)和γ-氨基丁酸(gaba)等5种神经递质相关。自上世纪50年代首次发现三环类抗抑郁药以来,已有多种抗抑郁药物被发现并广泛使用。目前临床使用的抗抑郁药物主要为:(1)单胺氧化酶抑制剂(maois),如吗氯贝胺(moclobemide);(2)三环类抗抑郁药(tcas),如地昔帕明(desipramine);(3)选择性5-羟色胺(5-ht)再摄取抑制剂(ssris),如氟西汀(fluoxetine)、帕罗西汀(paroxetine);(4)去甲肾上腺素(na)再摄取抑制剂(nris),如瑞波西汀(reboxitine);(5)去甲肾上腺素能和特异性5-ht再摄取抑制剂(ndris),如米氮平(mirtazapine);(6)5-ht和na双重再摄取抑制剂(snris),如文拉法辛(venlafaxine)和度洛西汀(duloxetine);(7)5-ht再吸收促进剂,如噻奈普汀(tianeptine)等。现有的抗抑郁药不能满足临床治疗需求,主要仍存在起效迟缓、不能有效改善认知损伤、性功能障碍、胃肠道不良反应、失眠及潜在的药物相互作用等缺陷,药物耐受性差。例如:选择性5-羟色胺(5-ht)再摄取抑制剂ssris存在起效延迟,通常在人体内数周之后才能产生抗抑郁疗效。而且,ssris对5-ht1和5-ht2受体作用不具有选择性,激活脊髓的5-ht1a受体有利于治疗抑郁症,但激活前脑的5-ht2受体可能导致激动或焦虑,因此ssris仍存在激动、焦虑、静坐不能和性功能障碍等不良反应。临床数据显示,ssris药物对约40%的患者没有明显抗抑郁疗效,而较新型的snris双重作用药物的疗效虽较ssris更加明显,但仍有相当数量的患者疗效不满意。此外,目前临床使用的多数抗抑郁药耐受性较差,仍存在诸多不良反应,例如:眩晕、呕吐、亢奋、心血管系统影响、性功能障碍等。5-ht1a受体激动剂可通过完全激动突触后膜的5-ht1a受体,负反馈抑制海马5-ht能神经元上的自身受体,减少投射到海马的中缝核5-ht能神经元在海马区释放神经递质发挥抗抑郁作用。5-ht1a受体激动剂可以克服大多数临床抗抑郁药物存在起效延迟的缺陷,并通过兴奋突触前后膜5-ht1a受体,减少不良反应。5-ht1a部分激动剂,同时激动突触前后膜的5-ht1a受体,对da能系统具有独特的双向效应:小剂量激动da受体,高剂量时则表现出da的受体拮抗作用。例如:丁螺环酮对5-ht1a受体具有很高的亲和性,作为选择性5-ht1a受体类抗抑郁药物,临床上用于广泛性焦虑障碍、抑郁的治疗。sigma-1受体在中枢神经系统和外周神经系统中均是高表达,主要分布于细胞膜,内质网和线粒体。中枢神经系统中sigma-1受体在神经保护,改善学习记忆,抗精神病、治疗药物成瘾及运动障碍等方面产生重要作用[psychopharmacology,2004,174(3):301.]。sigma-1受体蛋白是作为大脑内质网中唯一的结合位点,被认为对于认知障碍改善有效[currentpharmaceuticaldesign,2012,18(7):875-883.]。许多研究表明sigma-1受体在抑郁症及认知功能改善等方面具有重要作用[humanpsychopharmacologyclinical&experimental,2010,25(25):193-200.]。目前临床上开发的sigma-1受体激动剂库他美新,用于重度抑郁症的治疗。skuza等人报道[pharmacologicalreportspr,2007,59(6):773.]:强迫游泳实验中,对大鼠进行5-ht1a受体激动剂和sigma-1受体激动剂联合用药表现出抗抑郁活性,而给予5-ht1a受体拮抗剂或sigma-1受体拮抗剂,仅产生部分抵消作用。研究结果表明5-ht1a受体和sigma-1受体均参与大鼠的抗抑郁过程,双重激动剂具有配伍增效、扩大抗抑郁谱的作用,同时研究表明对药物快速起效和改善认知损伤具有重要意义。近年来,5-ht1a受体和sigma-1受体双重激动剂作为新型抗抑郁药物开发已成为该领域研究新的热点方向。例如:由大冢制药公司开发的抗抑郁药物opc-14523,目前处于临床试验,在体外实验中对5-ht1a受体,sigma-1受体和5-羟色胺转运体蛋白均表现出高亲和活性,但在体内抗抑郁实验中仅对5-ht1a受体和sigma-1受体产生作用,而无明显5-ht再摄取抑制作用,该药物相较于ssris,起效时间显著减少,此外,对于认知功能改善已表现出明显作用[journalofpharmacology&experimentaltherapeutics,2004,310(2):578.]。上述研究表明:5-ht1a受体激动剂具有快速抗抑郁作用,应答率高,且药物耐受性良好。sigma-1受体激动剂具有抗抑郁和认知改善作用,起效速度快。5-ht1a受体和sigma-1受体双重激动剂具有协同配伍增效、扩大抗抑郁谱作用,同时对药物快速起效和认知损伤改善具有重要作用。因此,进行作用于5-ht1a受体和sigma-1受体的双靶点新结构化合物的抗抑郁应用研究,已成为目前抗抑郁新药开发的重要研究方向,具有新颖性、创造性和重要科学价值。专利cn1434189a中公开了一类4-苯基-1-哌嗪基(哌啶基)和四氢吡啶基衍生物,对多巴胺d3、d4受体具有高亲和作用,可应用于治疗精神病的药物,代表化合物包括化合物349672-17-9和化合物349671-99-4,化学结构见图1.。该专利中公开了上述两个化合物对多巴胺d3、d4受体具有高亲和力,可用于治疗精神病,包括精神分裂症的阳性和阴性症状,情感性神经疾患,如一般性焦虑症、恐慌症和强迫行为与观念症,压抑,攻击性行为,认知障碍和由于用l-多巴治疗诱发的运动障碍。文献[chemmedchem,2010,5,1300-1317.]中报道了化合物1243651-06-0,其化学结构见附图1.,与多巴胺d2、d3受体具有高亲和力。本专利公开了一类苯骈氮杂烷基芳基哌嗪衍生物,发明人发现,所公开的化合物尽管在化学结构上与上述化合物存在相似之处,但亦存在结构特征的本质不同,这直接造成本发明专利化合物在药理作用机制和治疗适应症的本质不同。具体地说,本发明化合物四氢异喹啉结构中的氮原子保持了较强的碱性,体内可经质子化后,与sigma-1受体发生特异性高亲和作用,而专利cn1434189a和文献[chemmedchem,2010,5,1300-1317.]中报道的化合物结构中对应的氮原子,体内不能形成质子化,因此存在本质性的区别。更具体地说,化合物349672-17-9和349671-99-4与多巴胺d3、d4受体具有高亲和作用,可用于治疗精神病,包括精神分裂症的阳性和阴性症状,情感性神经疾患。化合物1243651-06-0,与多巴胺d3受体具有高亲和力。化合物与多巴胺系统d2、d3或d4受体高亲和作用,不可避免地导致多巴胺(dopamine,da)功能的平衡失调,引起较严重锥外体系的副作用(eps),例如:运动不能、肌张力高、震颤和自主神经功能紊乱等副作用。此外,体外试验表明化合物349672-17-9、349671-99-4和1243651-06-0均与sigma-1受体无亲和活性(见实施例25)。本专利公开的化合物通过与sigma-1受体和5-ht1a受体的双重高亲和作用,动物体内具有明显的快速抗抑郁作用,而且本专利公开的化合物与多巴胺d2、d3、d4受体均无亲和活性,例如:化合物iv-2、iv-4、iv-5(见实施例25、27)。而且动物体内试验中未见明显锥体外系副反应,毒副作用小,具有更高效、低毒的临床治疗应用前景。目前sigma-1受体蛋白结构结晶已公开[nature.2016,532(7600):527-30.],发明人采用计算机软件将化合物iv-5与sigma-1受体蛋白进行分子对接,发现化合物iv-5结构中的四氢异喹啉的碱性氮原子经质子化后,与sigma-1受体蛋白的氨基酸残基谷氨酸172和苯丙氨酸107相互作用,进一步证明,这是保持sigma-1受体高亲和作用的重要原因(示意图详见附图2.)。上述发现说明本发明的化合物结构与现有报道的化合物结构存在特性性的本质不同,因此其药理作用机制和治疗适应症的实质性特点。技术实现要素:本发明的目的是公开一种苯骈氮杂烷基芳基哌嗪衍生物及在制备药物中的应用,以克服现有技术存在的缺陷。本发明所述的苯骈氮杂烷基芳基哌嗪衍生物,为具有式(iv)所示的化合物或其游离碱或盐,盐为盐酸盐、二盐酸盐、溴氢酸盐、硫酸盐、三氟醋酸盐或甲磺酸盐等,优选的盐为盐酸盐、二盐酸盐、二溴氢酸盐,其盐可含0.5-3分子的结晶水:其中:r代表c1-c5的直链或支链的烷基,其中,烷基上的氢原子可任选被1-3个氟原子替代;r1代表h,och3、cl、ch3或cf3;r2代表h,cf3、cl或ch3;r3代表h,f或cl;r4、r5独立代表h,f、cl、och3或cn;x代表ch或n;n=0或1;m=0、1或2;优选的,r代表甲基、乙基、三氟甲基、正丙基或异丙基;r1代表h,och3、cl、ch3或cf3;r2代表h,cf3、cl或ch3;r3代表h,f或cl;x代表ch或n;n=0或1;m=0、1或2;优选的化合物包括:iv-12-(3-(4-(2,3-二甲基苯基)哌嗪-1-基)丙基)-1,2,3,4-四氢异喹啉、iv-22-(3-(4-(2,3-二氯苯基)哌嗪-1-基)丙基)-1,2,3,4-四氢异喹啉、iv-32-(2-(4-(2,3-二甲基苯基)哌嗪-1-基)乙基)-1,2,3,4-四氢异喹啉、iv-42-(2-(4-(2,3-二氯苯基)哌嗪-1-基)乙基)-1,2,3,4-四氢异喹啉、iv-56,7-二氯-2-(2-(4-(2,3-二氯苯基)哌嗪-1-基)乙基)-1,2,3,4-四氢异喹啉、iv-66,7-二氯-2-(3-(4-(2,3-二氯苯基)哌嗪-1-基)丙基)-1,2,3,4-四氢异喹啉、iv-76,7-二氯-2-(2-(4-(2,3-二氯苯基)哌嗪-1-基)乙基)-1-甲基-1,2,3,4-四氢异喹啉、iv-86,7-二氯-2-(3-(4-(2,3-二氯苯基)哌嗪-1-基)丙基)-1-甲基-1,2,3,4-四氢异喹啉、iv-96,7-二氯-2-(3-(4-(3-氯苯基)哌嗪-1-基)丙基)-1-甲基-1,2,3,4-四氢异喹啉、iv-106,7-二氯-2-(2-(4-(3-氯苯基)哌嗪-1-基)乙基)-1-甲基-1,2,3,4-四氢异喹啉、iv-116,7-二氯-2-(3-(4-(3-三氟甲基苯基)哌嗪-1-基)丙基)-1-甲基-1,2,3,4-四氢异喹啉、iv-126,7-二氯-2-(2-(4-(3-(三氟甲基苯基)哌嗪-1-基)乙基)-1-甲基-1,2,3,4-四氢异喹啉、iv-136,7-二氯-2-(3-(4-(4-氟-2-甲氧基苯基)哌嗪-1-基)丙基)-1-甲基-1,2,3,4-四氢异喹啉、iv-146,7-二氯-2-(2-(4-(4-氟-2-甲氧基苯基)哌嗪-1-基)乙基)-1-甲基-1,2,3,4-四氢异喹啉、iv-15(r)-8-氯-3-(2-(4-(2,3-二氯苯基)哌嗪-1-基)乙基)-1-甲基-2,3,4,5-四氢-1h-苯骈氮杂、iv-16(r)-8-氯-3-(3-(4-(2,3-二氯苯基)哌嗪-1-基)丙基)-1-甲基-2,3,4,5-四氢-1h-苯骈氮杂、iv-172-(3-(4-(2,3-二氯苯基)哌嗪-1-基)丙基)-1-甲基-1,2,3,4-四氢异喹啉-7-甲腈、iv-182-(2-(4-(2,3-二氯苯基)哌嗪-1-基)乙基)-1-甲基-1,2,3,4-四氢异喹啉-7-甲腈、iv-192-(2-(4-(2,3-二氯苯基)哌嗪-1-基)乙基)-1-甲基-1,2,3,4-四氢异喹啉、iv-202-(2-(4-(2-甲氧基苯基)哌嗪-1-基)乙基)-1-甲基-1,2,3,4-四氢异喹啉或具体的化学结构式如下表所示:其中,进一步优选的化合物包括:iv-56,7-二氯-2-(2-(4-(2,3-二氯苯基)哌嗪-1-基)乙基)-1,2,3,4-四氢异喹啉或iv-66,7-二氯-2-(3-(4-(2,3-二氯苯基)哌嗪-1-基)丙基)-1,2,3,4-四氢异喹啉。本发明化合物可采用如下合成路线合成,具体实验操作见本专利的实施例部分。首先,将相应的芳基哌嗪化合物(i)与1-溴-2-氯乙烷或1-溴-3-氯丙烷进行氮烷基化反应制备中间体(ii),再与关键中间体(iii)进行缩合反应,得目标新化合物(iv),最后经成盐制备相应的盐(v)。采用上述合成路线,可获得具体目标化合物iv-1至iv-18。所涉及的原料可采用商业化产品,其中部分关键中间体(iii)的合成路线如下。1、关键中间体的制备上述合成路线中关键中间体(iii)的化合物包括如下化合物结构,因此可具体分为iii-1、iii-2、iii-3三类:(1)以取代苯胺化合物iii-1-5为起始原料,在hbr/nano2作用下生成重氮盐,经麦尔外因芳基化反应与丙烯酸甲酯偶联得iii-1-4,再经锌粉脱溴,碱水解得化合物iii-1-3,然后在socl2/dmf作用下生成酰氯,与叠氮化钠作用经curtis反应生成相应的异氰酸酯中间体iii-1-2,经傅克酰基化得iii-1-1,最后经硼烷还原得到目标中间体iii-1,其合成路线如下:(2)以取代苯甲醛化合物iii-2-5为起始原料,首先与硝基甲烷缩合、脱水得化合物iii-2-4,经还原、酰氯化得iii-2-2,经三氯氧磷/五氧化二磷关环后,还原亚胺双键得目标中间体iii-2,具体合成路线如下:(3)中间体iii-3可参见专利wo200807111所报道的合成方法制备,以对氯苯乙酸为起始原料,与异丙醇胺反应得iii-3-4,经羰基还原、羟基氯化得iii-3-3,再经傅克反应关环得(r,s)-8-氯-1-甲基-2,3,4,5-四氢-1h-3-苯并氮杂,最后经l-酒石酸拆分,游离,成盐得目标中间体iii-3(合成路线如下)。亦或采用直接商业化购买。动物试验证明,所述的苯骈氮杂烷基芳基哌嗪衍生物,对于精神疾病具有显著的治疗效果,可以用于制备治疗精神疾病的药物,所述的精神疾病包括各种类型的抑郁症、焦虑症、各种双向情感障碍、精神分裂症、帕金森病(pd)、亨廷顿舞蹈病(hd)、阿尔茨海默氏病、老年性痴呆、阿尔茨海默氏型痴呆、记忆障碍、执行功能丧失、血管性痴呆和其它痴呆,以及与智力、学习或记忆相关的功能障碍性疾病。动物试验同时证明,所述的苯骈氮杂烷基芳基哌嗪衍生物对疼痛具有明显的镇痛作用,可以用于制备治疗疼痛疾病的药物;所述的疼痛疾病,包括各种伤害感受性疼痛、急性疼痛、慢性疼痛、神经性疼痛、精神性疼痛和混合性疼痛等。它特别包括但不限于:手术后疼痛、神经原性疼痛、中枢性痛、躯体痛、内脏痛、慢性后背痛、颈和腰痛、癌症痛、炎症疼痛、糖尿病性神经痛、坐骨神经痛、紧张性头痛、丛集性头痛、每日慢性头痛、疱疹神经痛、面部和口腔神经痛以及肌筋膜痛综合征、假性肢痛、残肢痛和截瘫痛、牙痛、耐阿片样物质疼痛、包括心脏手术和乳房切除术在内的术后疼痛、心绞痛、骨盆疼痛、膀胱炎以及阴道前庭炎和睾丸痛在内的泌尿生殖道疼痛、月经前期疼痛综合症、中风后疼痛、过敏性肠综合征、劳累和分娩疼痛、分娩后疼痛、因烧伤和化学损伤或日晒导致的疼痛和骨损伤性疼痛。本发明发现苯骈氮杂烷基芳基哌嗪衍生物在体外5-ht1a和sigma-1受体亲和试验中,表现出与5-ht1a和sigma-1受体的高亲和活性。动物模型研究结果表明:化合物iv-5二盐酸盐在小鼠尾悬挂试验中,经灌胃给药,三组剂量下均具有明显抗抑郁作用,其口服吸收良好;化合物iv-6二盐酸盐在小鼠尾悬挂试验中,经灌胃给药,三组剂量下均具有明显抗抑郁作用,其口服吸收良好;化合物iv-5和iv-6的二盐酸盐ames试验均呈阴性。化合物iv-5和iv-6可作为新型抗抑郁新药开发。本发明的衍生物可以组合物的形式通过口服、注射等方式施用于需要这种治疗的患者。给药剂量一般为0.02~5mg/kg(口服)或0.01~2mg/kg(注射),具体可根据临床实验结果及患者的病情、年龄等由医师决定。所述组合物,包括治疗治疗有效量所述的苯骈氮杂烷基芳基哌嗪衍生物和医药学上可接受的载体,所述及的载体是指药学领域常规的载体,例如:稀释剂、赋形剂如水等;粘合剂如纤维素衍生物、明胶、聚乙烯吡咯烷酮等;填充剂如淀粉等;崩裂剂如碳酸钙、碳酸氢钠;润滑剂如硬脂酸钙或硬脂酸镁等。另外,还可以在组合物中加入其他辅助剂如香味剂和甜味剂。用于口服时,可将其制备成常规的固体制剂如片剂、粉剂或胶囊等;用于注射时,可将其制备成注射液。本发明的组合物的各种剂型可以采用医学领域常规的方法进行制备,其中活性成分的含量为0.1%~99.5%(重量)。本发明人发现,本发明的苯骈氮杂烷基芳基哌嗪衍生物口服吸收好、起效快、改善认知损伤良好、毒副作用小,能够扩大临床抗抑郁药物的应用选择,满足抑郁病人药物治疗的需要。本发明所述的苯骈氮杂烷基芳基哌嗪衍生物,显示出对中枢神经系统的作用,特别是对5-ht1a受体和sigma-1受体的高活性,可通过作用于5-ht1a受体发挥多种生理和药理作用,可通过作用sigma-1受体发挥多种生理和药理作用,特别是产生快速抗抑郁作用和修复认知损伤作用,特别是用于抗抑郁、改善认知损伤、抗双向情感障碍、抗焦虑、抗精神分裂症、抗精神类疾病,还可以用作制备其它药物活性化合物的中间体。本发明所述的苯骈氮杂烷基芳基哌嗪衍生物具有非常有用的药学性质及良好的耐受性,尤其是作为新型抗抑郁、抗焦虑药物的应用。该类化合物为新型5-ht1a受体和sigma-1受体的双靶点活性作用抗抑郁剂。该类化合物还具有较小的毒副作用和较高的安全指数。附图说明:图1:化合物349671-99-4、349672-17-9和1243651-06-0的化学结构图2:化合物iv-5与sigma-1受体蛋白进行分子对接图具体实施方式1、中间体化合物iii-1的制备中间体iii-1-4:2-溴-3-(3,4-二氯苯基)丙酸甲酯的制备将48%的溴化氢16ml加入到丙酮溶液100ml中,缓慢加入3,4-二氯苯胺(8.1g,50mmol),保持内温-5℃缓慢滴加亚硝酸钠4.2g的水溶液100ml,30min内滴毕。然后向其中加入丙烯酸甲酯43g和cubr粉末7.5mg,撤去冰浴,待温度缓慢上升至25℃后搅拌30min,蒸出溶剂,以水和二氯甲烷各50ml分配,二氯甲烷层以水反洗,无水硫酸镁干燥,过滤,滤液蒸干得产物14.2g。中间体iii-1-3:3,4-二氯苯丙酸的制备将2-溴-3-(3,4-二氯苯基)丙酸甲酯13.1g溶于80ml冰醋酸中,缓慢加入锌粉5.24g,加毕后搅拌30min,滤出锌粉,减压蒸馏除去乙酸,以二氯甲烷和水各50ml分配,有机层用水洗,饱和食盐水洗,无水硫酸钠干燥,过滤,滤液浓缩,得到红棕色油状物,将此油状物溶于5%naoh溶液50ml中,回流2h,室温冷却,沉淀析出,过滤,滤液加5%hcl调节至酸性,逐渐析出浅黄色固体,过滤,低温干燥得3,4-二氯苯丙酸3.5g。esi-ms[m+h]+:m/z219.0中间体iii-1-2:1,2-二氯-4-(2-异氰酸乙基)苯的制备将3,4-二氯苯丙酸1.1g(5mmol)和1滴dmf加入到5ml甲苯中,冰浴下缓慢滴加二氯亚砜0.75g(6.5mmol),55-60℃条件下搅拌2.5h,冷却,蒸出甲苯,得到3,4-二氯本丙酰氯1.2g。将酰氯溶于15ml丙酮中,保持内温低于0℃,滴加nan30.91g(14mmol)的水溶液5ml,搅拌5分钟,倒入20g冰水混合物中,甲苯萃取,低温下无水硫酸镁干燥,过滤,60℃下缓慢蒸出甲苯,得到目标产物900mg。中间体iii-1-1:6,7-二氯-3,4-二氢异喹啉-1(2h)-酮的制备向100ml四氯乙烯中缓慢加入1.4galcl3,将1,2-二氯-4-(2-异氰酸乙基)苯900mg的5ml四氯乙烯溶液缓慢滴入,70-75℃反应3h,冷却,冰浴条件下滴加3nhcl溶液,调节ph至3-4,有固体析出,过滤,固体用二氯甲烷溶解,加水萃取,有机相加无水硫酸钠干燥,浓缩,得黄色固体700mg。中间体iii-1:6,7-二氯-1,2,3,4-四氢异喹啉的制备取6,7-二氯-3,4-二氢异喹啉-1(2h)-酮2.8g,n2保护下加40ml四氢呋喃,搅拌5min,加无水2mbh3/thf23ml,加完回流18h,缓慢滴加水2.5ml,再加入12ml3nhcl溶液,固体析出,过滤,40℃真空干燥,得到白色固体1.2g。esi-ms[m+h]+:m/z202.12、中间体化合物iii-2的制备中间体iii-2-4:(e)-1,2-二氯-4-(2-硝基乙烯基)苯的制备将3,4-二氯苯甲醛(15.0g,0.086mol),硝基甲烷(105.3g,1.72mol),醋酸铵nh4oac(13.3g,0.172mol)和乙酸(51.8g,0.862mol)依次加入到500ml的三口烧瓶中,在68℃条件下反应6h。反应结束后,将反应液倒入100ml乙酸乙酯和100ml水中,分离有机相,饱和碳酸氢钠洗涤至ph=7,饱和食盐水洗涤,浓缩得到20.8g淡黄色固体,无水乙醇重结晶。中间体iii-2-3:2-(3,4-二氯苯基)乙胺的制备0℃条件下,将化合物iii-2-4(6.0g,27.68mmol)的无水thf溶液(81ml)溶液滴加到lialh4(3.2g,82.96mmol)的无水et2o(89ml)中,一小时内滴加完毕,升至室温,搅拌反应6h。反应结束后,用十水硫酸钠淬灭未反应的lialh4,过滤,滤液浓缩,草酸成盐纯化,得到6.83g黄棕色固体。中间体iii-2-2:n-(3,4-二氯苯乙基)乙酰胺的制备化合物iii-2-3(4.26g,22.22mmol)、二氯甲烷(25ml)和三乙胺(2.47g,24.44mol)加至装有干燥管和温度计的150ml三颈瓶中,控温0℃以下,缓慢滴加乙酰氯(1.92g,24.44mol),滴毕升至室温续搅30min,静置分层,水层用二氯甲烷(10ml×3)洗涤,合并有机相,用水(30ml)洗涤,无水硫酸钠干燥,抽滤,滤液减压下蒸除溶剂,剩余浅黄色油状物,硅胶柱层析得到2.4g黄色蜡状固体。中间体iii-2-1:6,7-二氯-1-甲基-3,4-二氢异喹啉的制备化合物iii-2-2(2.61g,11.29mmol)、pocl3(20ml,124mmol)和p2o5(0.53g,3.76mmol)加至装有干燥管和温度计的100ml三颈瓶中,缓慢升温至回流3h,冷却至室温,减压蒸除未反应的pocl3,加入碎冰(50g),搅拌下加饱和氢氧化钠溶液调至ph9,加入乙酸乙酯(100ml),静置分层,水层用乙酸乙酯(15ml×3)洗涤,合并有机相,无水硫酸镁干燥,抽滤,滤液蒸除溶剂,得浅棕红色油状1.9g。中间体iii-2:6,7-二氯-1-甲基-1,2,3,4-四氢异喹啉的制备将化合物iii-2-1(1.90g,8.92mmol)和无水乙醇(20ml)加至100ml三颈瓶中,搅拌下缓慢分次加入nabh4(0.39g,9.81mmol),室温反应1.5h,减压蒸除溶剂,剩余物中加入水(20ml),加饱和氢氧化钠溶液调至ph=11,加入乙酸乙酯(10ml),静置分层。水层用乙酸乙酯(10ml×3)洗涤,合并有机相,无水硫酸钠干燥,抽滤,滤液减压蒸除溶剂,得浅棕红色油状1.5g。esi-ms[m+h]+:m/z216.03、目标化合物ⅲ-1~ⅲ-18的合成通法:取化合物(i)的盐(0.1mol,1.0eq.)加二氯甲烷150ml溶解,将naoh水溶液(5.2g,50ml)滴加至上述溶液中,室温搅拌0.5h,静置分层,水相30ml二氯甲烷萃取,合并有机相,浓缩得油状物。将上述油状物溶解于30ml丙酮中,加入1-溴-3-氯丙烷(或)1-溴-2-氯乙烷(0.11mol,1.1equiv.),再加25%naoh水溶液18ml,室温搅拌反应12h停止搅拌,静置分层,水层二氯甲烷萃取,合并有机相,饱和食盐水洗涤,分液,有机相经无水硫酸钠干燥,过滤,浓缩,硅胶柱层析纯化(洗脱剂:二氯甲烷),浓缩,得中间体(ii),类白色固体。取中间体(ii)(4.8mmol,1.0eq.),中间体(iii)(4.8mmol,1.0eq.),n,n-二异丙基乙胺2.48g(19.2mmol,4.0eq.),碘化钾0.8g(4.8mmol,1.0eq.)溶于30ml乙腈,升温回流,搅拌6.5h,tlc示原料基本反应完全,停止加热,减压蒸馏除去溶剂,加入水、乙酸乙酯各100ml,静置分层,水层经乙酸乙酯萃取3次,合并有机相,饱和食盐水洗涤,无水硫酸钠干燥,浓缩后经硅胶柱层析纯化得目标化合物(iv),加30ml乙酸乙酯溶解,滴加3mhcl/etoac调节ph=2~3,固体析出,过滤,真空干燥,得目标化合物二盐酸盐(v),白色固体。具体实施例实施例12-(3-(4-(2,3-二甲基苯基)哌嗪-1-基)丙基)-1,2,3,4-四氢异喹啉(iv-1)二盐酸盐的制备及2-(3-(4-(2,3-二甲基苯基)哌嗪-1-基)丙基)-1,2,3,4-四氢异喹啉(iv-1)氢溴酸盐的制备取化合物1-(2,3-二甲基苯基)哌嗪盐酸盐(0.1mol,1.0eq.)加二氯甲烷150ml溶解,将naoh水溶液(5.2g,50ml)滴加至上述溶液中,室温搅拌0.5h,静置分层,水相30ml二氯甲烷萃取,合并有机相,浓缩得油状物,溶解于30ml丙酮中,加入1-溴-3-氯丙烷(0.11mol,1.1equiv.),再加25%naoh水溶液18ml,室温搅拌反应12h停止搅拌,静置分层,水层二氯甲烷萃取,合并有机相,饱和食盐水洗涤,分液,有机相经无水硫酸钠干燥,过滤,浓缩,硅胶柱层析纯化(洗脱剂:二氯甲烷),浓缩,得1-(3-氯丙基)-4-(2,3-二甲基苯基)哌嗪,类白色固体14.2g。esi-ms[m+h]+:m/z267.1取1-(3-氯丙基)-4-(2,3-二甲基苯基)哌嗪(4.8mmol,1.0eq.),1,2,3,4-四氢异喹啉(4.8mmol,1.0eq.),n,n-二异丙基乙胺2.48g(19.2mmol,4.0eq.),碘化钾0.8g(4.8mmol,1.0eq.)溶于30ml乙腈,升温回流,搅拌6.5h。按合成通法进行后处理操作,得白色固体,目标化合物(iv-1)二盐酸盐,收率85%。hplc:99.74%。元素分析:c24h33n3·2hcl(理论值%:c66.04,h8.08,n9.63,cl16.25;实验值%c66.23,h8.15,n9.78,cl16.06)esi-ms[m+h]+:m/z364.21h-nmr(400mhz),dmso-d6:δ11.36(s,2h),7.32-7.18(m,4h),7.09(t,j=8.0hz,1h),6.94(d,j=8.4hz,1h),6.91(d,j=8.4hz,1h),4.60(d,j=14.8hz,1h),4.31(dd,j=15.4,8.0hz,1h),3.90-3.70(m,8h),3.68-3.50(m,2h),3.41-3.32(m,4h),3.28-3.01(m,2h),2.45-2.30(m,2h),2.23(s,3h),2.18(s,3h).取1-(3-氯丙基)-4-(2,3-二甲基苯基)哌嗪(4.8mmol,1.0eq.),1,2,3,4-四氢异喹啉(4.8mmol,1.0eq.),n,n-二异丙基乙胺2.48g(19.2mmol,4.0eq.),碘化钾0.8g(4.8mmol,1.0eq.)溶于30ml乙腈,升温回流,搅拌6.5h。按合成通法进行后处理操作得到目标化合物iv-1,加30ml丙酮溶解,滴加40%氢溴酸水溶液调节ph=2~3,搅拌下固体析出,过滤,真空干燥,得目标化合物iv-1的氢溴酸酸盐,收率45%。元素分析:c24h33n3·2hbr(理论值%:c54.87,h6.71,n8.00,br30.42;实验值%c55.03,h6.65,n8.12,br30.26)实施例22-(3-(4-(2,3-二氯苯基)哌嗪-1-基)丙基)-1,2,3,4-四氢异喹啉(iv-2)二盐酸盐的制备取化合物1-(2,3-二氯苯基)哌嗪盐酸盐(0.1mol,1.0eq.),加二氯甲烷150ml溶解,将naoh水溶液(5.2g,50ml)滴加至上述溶液中,室温搅拌0.5h,静置分层,水相30ml二氯甲烷萃取,合并有机相,浓缩得油状物,溶解于30ml丙酮中,加入1-溴-3-氯丙烷(0.11mol,1.1equiv.),再加25%naoh水溶液18ml,室温搅拌反应12h停止搅拌,静置分层,水层二氯甲烷萃取,合并有机相,饱和食盐水洗涤,分液,有机相经无水硫酸钠干燥,过滤,浓缩,硅胶柱层析纯化(洗脱剂:二氯甲烷),浓缩,得1-(3-氯丙基)-4-(2,3-二氯苯基)哌嗪,类白色固体15.3g。esi-ms[m+h]+:m/z307.1取1-(3-氯丙基)-4-(2,3-二氯苯基)哌嗪(4.8mmol,1.0eq.),1,2,3,4-四氢异喹啉(4.8mmol,1.0eq.),n,n-二异丙基乙胺2.48g(19.2mmol,4.0eq.),碘化钾0.8g(4.8mmol,1.0eq.)溶于30ml乙腈,升温回流,搅拌6.5h。按合成通法进行后处理操作,得白色固体,目标化合物(iv-2)二盐酸盐,收率78%。hplc:99.76%。esi-ms[m+h]+:m/z404.11h-nmr(400mhz),dmso-d6:δ11.56(s,1h),11.33(s,1h),7.42-7.34(m,2h),7.30-7.19(m,5h),4.60(d,j=15.6hz,1h),4.31(dd,j=15.6,7.6hz,1h),3.78-3.60(m,3h),3.49-3.41(m,2h),3.37-3.20(m,10h),3.04(m,1h),2.45-2.32(m,2h).实施例32-(2-(4-(2,3-二甲基苯基)哌嗪-1-基)乙基)-1,2,3,4-四氢异喹啉(iv-3)二盐酸盐的制备取化合物1-(2,3-二甲基苯基)哌嗪盐酸盐(0.1mol,1.0eq.)加二氯甲烷150ml溶解,将naoh水溶液(5.2g,50ml)滴加至上述溶液中,室温搅拌0.5h,静置分层,水相30ml二氯甲烷萃取,合并有机相,浓缩得油状物,溶解于30ml丙酮中,加入1-溴-2-氯乙烷(0.11mol,1.1equiv.),再加25%naoh水溶液18ml,室温搅拌反应12h停止搅拌,静置分层,水层二氯甲烷萃取,合并有机相,饱和食盐水洗涤,分液,有机相经无水硫酸钠干燥,过滤,浓缩,硅胶柱层析纯化(洗脱剂:二氯甲烷),浓缩,得1-(2-氯乙基)-4-(2,3-二甲基苯基)哌嗪,类白色固体13.5g。esi-ms[m+h]+:m/z253.1取1-(2-氯乙基)-4-(2,3-二甲基苯基)哌嗪(4.8mmol,1.0eq.),1,2,3,4-四氢异喹啉(4.8mmol,1.0eq.),n,n-二异丙基乙胺2.48g(19.2mmol,4.0eq.),碘化钾0.8g(4.8mmol,1.0eq.)溶于30ml乙腈,升温回流,搅拌6.5h。按合成通法进行后处理操作,得白色固体,目标化合物(iv-3)二盐酸盐,收率81%。hplc:98.71%。esi-ms[m+h]+:m/z350.21h-nmr(400mhz),dmso-d6:δ11.06(s,2h),7.35-7.24(m,3h),7.24-7.08(m,1h),7.08(t,j=7.6hz,1h),6.94(dd,j=7.6hz,1h),6.91(dd,j=8.0hz,1h),4.80-4.60(m,1h),4.49-4.25(m,1h),3.95-3.65(m,6h),3.40-3.00(m,10h),2.23(s,3h),2.19(s,3h).实施例42-(2-(4-(2,3-二氯苯基)哌嗪-1-基)乙基)-1,2,3,4-四氢异喹啉(iv-4)二盐酸盐的制备取化合物1-(2,3-二氯苯基)哌嗪盐酸盐(0.1mol,1.0eq.)加二氯甲烷150ml溶解,将naoh水溶液(5.2g,50ml)滴加至上述溶液中,室温搅拌0.5h,静置分层,水相30ml二氯甲烷萃取,合并有机相,浓缩得油状物,溶解于30ml丙酮中,加入1-溴-2-氯乙烷(0.11mol,1.1equiv.),再加25%naoh水溶液18ml,室温搅拌反应12h停止搅拌,静置分层,水层二氯甲烷萃取,合并有机相,饱和食盐水洗涤,分液,有机相经无水硫酸钠干燥,过滤,浓缩,硅胶柱层析纯化(洗脱剂:二氯甲烷),浓缩,得1-(2-氯乙基)-4-(2,3-二氯苯基)哌嗪,类白色固体14.5g。esi-ms[m+h]+:m/z293.0取1-(2-氯乙基)-4-(2,3-二氯苯基)哌嗪(4.8mmol,1.0eq.),1,2,3,4-四氢异喹啉(4.8mmol,1.0eq.),n,n-二异丙基乙胺2.48g(19.2mmol,4.0eq.),碘化钾0.8g(4.8mmol,1.0eq.)溶于30ml乙腈,升温回流,搅拌6.5h。按合成通法进行后处理操作,得白色固体,目标化合物(iv-4)二盐酸盐,收率80%。hplc:99.19%。esi-ms[m+h]+:m/z390.11h-nmr(400mhz),dmso-d6:δ10.89(s,2h),7.34-7.27(m,2h),7.26-7.18(m,3h),7.17-7.11(m,2h),4.80-4.58(m,1h),4.49-4.25(m,1h),3.95-2.96(m,16h).实施例56,7-二氯-2-(2-(4-(2,3-二氯苯基)哌嗪-1-基)乙基)-1,2,3,4-四氢异喹啉(iv-5)二盐酸盐的制备取1-(2-氯乙基)-4-(2,3-二氯苯基)哌嗪(4.8mmol,1.0eq.),6,7-二氯-1,2,3,4-四氢异喹啉(4.8mmol,1.0eq.),n,n-二异丙基乙胺2.48g(19.2mmol,4.0eq.),碘化钾0.8g(4.8mmol,1.0eq.)溶于30ml乙腈,升温回流,搅拌6.5h。按合成通法进行后处理操作,得白色固体,目标化合物(iv-5)二盐酸盐,收率74%。元素分析:c21h23cl4n3·2hcl(理论值%:c,47.40,h4.74,n7.90,cl39.97;实验值%c47.33,h4.67,n7.88,cl39.79)esi-ms[m+h]+:m/z458.11h-nmr(400mhz,dmso)δ11.19(s,2h),7.62(s,1h),7.55(s,1h),7.39(dd,j=8.0,2.0hz,1h),7.36(t,j=8.4hz,1h),7.23(dd,j=7.6,2.0hz,1h),4.70-4.56(m,1h),4.45-4.24(m,1h),4.05-2.96(m,16h).实施例66,7-二氯-2-(3-(4-(2,3-二氯苯基)哌嗪-1-基)丙基)-1,2,3,4-四氢异喹啉(iv-6)二盐酸盐的制备取1-(3-氯丙基)-4-(2,3-二氯苯基)哌嗪(4.8mmol,1.0eq.),6,7-二氯-1,2,3,4-四氢异喹啉(4.8mmol,1.0eq.),n,n-二异丙基乙胺2.48g(19.2mmol,4.0eq.),碘化钾0.8g(4.8mmol,1.0eq.)溶于30ml乙腈,升温回流,搅拌6.5h。按合成通法进行后处理操作,得白色固体,目标化合物(iv-6)二盐酸盐,收率71%。esi-ms[m+h]+:m/z472.11h-nmr(400mhz),dmso-d6:δ11.32(s,1h),11.30(s,1h),7.62(s,1h),7.56(s,1h),7.39(dd,j=7.6,2.0hz,1h),7.37(t,j=8.0hz,1h),7.23(dd,j=7.6,2.0hz,1h),4.60(d,j=14.8hz,1h),4.41-4.29(m,1h),3.80-3.69(m,1h),3.68-3.55(m,2h),3.54-3.39(m,6h),3.37-3.12(m,6h),3.12-3.01(m,1h),2.42-2.25(m,2h).实施例76,7-二氯-2-(2-(4-(2,3-二氯苯基)哌嗪-1-基)乙基)-1-甲基-1,2,3,4-四氢异喹啉(iv-7)二盐酸盐的制备取1-(2-氯乙基)-4-(2,3-二氯苯基)哌嗪(4.8mmol,1.0eq.),6,7-二氯-1-甲基-1,2,3,4-四氢异喹啉(4.8mmol,1.0eq.),n,n-二异丙基乙胺2.48g(19.2mmol,4.0eq.),碘化钾0.8g(4.8mmol,1.0eq.)溶于30ml乙腈,升温回流,搅拌6.5h。按合成通法进行后处理操作,得白色固体,目标化合物(iv-7)二盐酸盐,收率79%。esi-ms[m+h]+:m/z472.11h-nmr(400mhz,dmso)δ11.32(b,2h),7.63(s,1h),7.56(s,1h),7.39(dd,1h),7.36(t,1h),7.23(dd,1h),4.96-4.61(m,1h),4.04-2.95(m,16h),1.75-1.50(m,3h).实施例86,7-二氯-2-(3-(4-(2,3-二氯苯基)哌嗪-1-基)丙基)-1-甲基-1,2,3,4-四氢异喹啉(iv-8)二盐酸盐的制备取1-(3-氯丙基)-4-(2,3-二氯苯基)哌嗪(4.8mmol,1.0eq.),6,7-二氯-1-甲基-1,2,3,4-四氢异喹啉(4.8mmol,1.0eq.),n,n-二异丙基乙胺2.48g(19.2mmol,4.0eq.),碘化钾0.8g(4.8mmol,1.0eq.)溶于30ml乙腈,升温回流,搅拌6.5h。按合成通法进行后处理操作,得白色固体,目标化合物(iv-8)二盐酸盐,收率76%。esi-ms[m+h]+:m/z486.11h-nmr(400mhz,dmso)δ11.48-11.21(m,2h),7.62(s,1h),7.56(s,1h),7.39(dd,1h),7.37(t,1h),7.23(dd,1h),4.94-4.60(m,1h),3.96-3.39(m,10h),3.36-2.21(m,8h),1.75-1.50(m,3h).实施例96,7-二氯-2-(3-(4-(3-氯苯基)哌嗪-1-基)丙基)-1-甲基-1,2,3,4-四氢异喹啉(iv-9)二盐酸盐的制备取化合物1-(3-氯苯基)哌嗪盐酸盐(0.1mol,1.0eq.)加二氯甲烷150ml溶解,将naoh水溶液(5.2g,50ml)滴加至上述溶液中,室温搅拌0.5h,静置分层,水相30ml二氯甲烷萃取,合并有机相,浓缩得油状物,溶解于30ml丙酮中,加入1-溴-3-氯丙烷(0.11mol,1.1eq.),再加25%naoh水溶液18ml,室温搅拌反应12h停止搅拌,静置分层,水层二氯甲烷萃取,合并有机相,饱和食盐水洗涤,分液,有机相经无水硫酸钠干燥,过滤,浓缩,硅胶柱层析纯化(洗脱剂:二氯甲烷),浓缩,得1-(3-氯丙基)-4-(3-氯苯基)哌嗪,类白色固体14.2g。esi-ms[m+h]+:m/z273.1取1-(3-氯丙基)-4-(3-氯苯基)哌嗪(4.8mmol,1.0eq.),6,7-二氯-1-甲基-1,2,3,4-四氢异喹啉(4.8mmol,1.0eq.),n,n-二异丙基乙胺2.48g(19.2mmol,4.0eq.),碘化钾0.8g(4.8mmol,1.0eq.)溶于30ml乙腈,升温回流,搅拌6.5h。按合成通法进行后处理操作,得白色固体,目标化合物(iv-9)二盐酸盐,收率73%。esi-ms[m+h]+:m/z452.11h-nmr(400mhz,dmso)δ11.46-11.20(m,2h),7.62(s,1h),7.55(s,1h),7.25(t,1h),7.04(s,1h),6.95(d,1h),6.85(d,1h),4.95-4.60(m,1h),3.96-3.39(m,10h),3.36-2.20(m,8h),1.76-1.52(m,3h).实施例106,7-二氯-2-(2-(4-(3-氯苯基)哌嗪-1-基)乙基)-1-甲基-1,2,3,4-四氢异喹啉(iv-10)二盐酸盐的制备取化合物1-(3-氯苯基)哌嗪盐酸盐(0.1mol,1.0eq.)加二氯甲烷150ml溶解,将naoh水溶液(5.2g,50ml)滴加至上述溶液中,室温搅拌0.5h,静置分层,水相30ml二氯甲烷萃取,合并有机相,浓缩得油状物,溶解于30ml丙酮中,加入1-溴-2-氯乙烷(0.11mol,1.1eq.),再加25%naoh水溶液18ml,室温搅拌反应12h停止搅拌,静置分层,水层二氯甲烷萃取,合并有机相,饱和食盐水洗涤,分液,有机相经无水硫酸钠干燥,过滤,浓缩,硅胶柱层析纯化(洗脱剂:二氯甲烷),浓缩,得1-(2-氯乙基)-4-(3-氯苯基)哌嗪,类白色固体14.2g。esi-ms[m+h]+:m/z259.1取1-(2-氯乙基)-4-(3-氯苯基)哌嗪(4.8mmol,1.0eq.),6,7-二氯-1-甲基-1,2,3,4-四氢异喹啉(4.8mmol,1.0eq.),n,n-二异丙基乙胺2.48g(19.2mmol,4.0eq.),碘化钾0.8g(4.8mmol,1.0eq.)溶于30ml乙腈,升温回流,搅拌6.5h。按合成通法进行后处理操作,得白色固体,目标化合物(iv-10)二盐酸盐,收率73%。esi-ms[m+h]+:m/z438.11h-nmr(400mhz,dmso)δ11.45-11.21(m,2h),7.63(s,1h),7.56(s,1h),7.26(t,1h),7.03(s,1h),6.97(d,1h),6.85(d,1h),4.96-4.60(m,1h),4.03-2.95(m,16h),1.76-1.53(m,3h).实施例116,7-二氯-2-(3-(4-(3-三氟甲基苯基)哌嗪-1-基)丙基)-1-甲基-1,2,3,4-四氢异喹啉(iv-11)二盐酸盐的制备取化合物1-(3-三氟甲基苯基)哌嗪盐酸盐(0.1mol,1.0eq.)加二氯甲烷150ml溶解,将naoh水溶液(5.2g,50ml)滴加至上述溶液中,室温搅拌0.5h,静置分层,水相30ml二氯甲烷萃取,合并有机相,浓缩得油状物,溶解于30ml丙酮中,加入1-溴-3-氯丙烷(0.11mol,1.1eq.),再加25%naoh水溶液18ml,室温搅拌反应12h停止搅拌,静置分层,水层二氯甲烷萃取,合并有机相,饱和食盐水洗涤,分液,有机相经无水硫酸钠干燥,过滤,浓缩,硅胶柱层析纯化(洗脱剂:二氯甲烷),浓缩,得1-(3-氯丙基)-4-(3-三氟甲基苯基)哌嗪,类白色固体15.7g。esi-ms[m+h]+:m/z307.1取1-(3-氯丙基)-4-(3-三氟甲基苯基)哌嗪(4.8mmol,1.0eq.),6,7-二氯-1-甲基-1,2,3,4-四氢异喹啉(4.8mmol,1.0eq.),n,n-二异丙基乙胺2.48g(19.2mmol,4.0eq.),碘化钾0.8g(4.8mmol,1.0eq.)溶于30ml乙腈,升温回流,搅拌6.5h。按合成通法进行后处理操作,得白色固体,目标化合物(iv-11)二盐酸盐,收率75%。esi-ms[m+h]+:m/z486.21h-nmr(400mhz,dmso)δ11.44-11.20(m,2h),7.62(s,1h),7.55(s,1h),7.47(t,1h),7.30-7.25(m,2h),7.15(d,1h),4.94-4.60(m,1h),3.97-3.39(m,10h),3.30-2.21(m,8h),1.75-1.52(m,3h).实施例126,7-二氯-2-(2-(4-(3-(三氟甲基苯基)哌嗪-1-基)乙基)-1-甲基-1,2,3,4-四氢异喹啉(iv-12)二盐酸盐的制备取化合物1-(3-三氟甲基苯基)哌嗪盐酸盐(0.1mol,1.0eq.)加二氯甲烷150ml溶解,将naoh水溶液(5.2g,50ml)滴加至上述溶液中,室温搅拌0.5h,静置分层,水相30ml二氯甲烷萃取,合并有机相,浓缩得油状物,溶解于30ml丙酮中,加入1-溴-2-氯乙烷(0.11mol,1.1eq.),再加25%naoh水溶液18ml,室温搅拌反应12h停止搅拌,静置分层,水层二氯甲烷萃取,合并有机相,饱和食盐水洗涤,分液,有机相经无水硫酸钠干燥,过滤,浓缩,硅胶柱层析纯化(洗脱剂:二氯甲烷),浓缩,得1-(2-氯乙基)-4-(3-三氟甲基苯基)哌嗪,类白色固体14.7g。esi-ms[m+h]+:m/z293.1取1-(2-氯乙基)-4-(3-三氟甲基苯基)哌嗪(4.8mmol,1.0eq.),6,7-二氯-1-甲基-1,2,3,4-四氢异喹啉(4.8mmol,1.0eq.),n,n-二异丙基乙胺2.48g(19.2mmol,4.0eq.),碘化钾0.8g(4.8mmol,1.0eq.)溶于30ml乙腈,升温回流,搅拌6.5h。按合成通法进行后处理操作,得白色固体,目标化合物(iv-12)二盐酸盐,收率74%。esi-ms[m+h]+:m/z472.21h-nmr(400mhz,dmso)δ11.47-11.19(m,2h),7.63(s,1h),7.56(s,1h),7.47(t,1h),7.29(d,1h),7.26(s,1h),7.14(d,1h),4.95-4.65(m,1h),4.03-2.93(m,16h),1.75-1.51(m,3h).实施例136,7-二氯-2-(3-(4-(4-氟-2-甲氧基苯基)哌嗪-1-基)丙基)-1-甲基-1,2,3,4-四氢异喹啉(iv-13)二盐酸盐的制备取化合物1-(4-氟-2-甲氧基苯基)哌嗪盐酸盐(0.1mol,1.0eq.)加二氯甲烷150ml溶解,将naoh水溶液(5.2g,50ml)滴加至上述溶液中,室温搅拌0.5h,静置分层,水相30ml二氯甲烷萃取,合并有机相,浓缩得油状物,溶解于30ml丙酮中,加入1-溴-3-氯丙烷(0.11mol,1.1eq.),再加25%naoh水溶液18ml,室温搅拌反应12h停止搅拌,静置分层,水层二氯甲烷萃取,合并有机相,饱和食盐水洗涤,分液,有机相经无水硫酸钠干燥,过滤,浓缩,硅胶柱层析纯化(洗脱剂:二氯甲烷),浓缩,得1-(3-氯丙基)-4-(4-氟-2-甲氧基苯基)哌嗪,类白色固体14.4g。esi-ms[m+h]+:m/z287.1取1-(3-氯丙基)-4-(4-氟-2-甲氧基苯基)哌嗪(4.8mmol,1.0eq.),6,7-二氯-1-甲基-1,2,3,4-四氢异喹啉(4.8mmol,1.0eq.),n,n-二异丙基乙胺2.48g(19.2mmol,4.0eq.),碘化钾0.8g(4.8mmol,1.0eq.)溶于30ml乙腈,升温回流,搅拌6.5h。按合成通法进行后处理操作,得白色固体,目标化合物(iv-11)二盐酸盐,收率75%。esi-ms[m+h]+:m/z466.21h-nmr(400mhz,dmso)δ11.43-11.20(m,2h),7.62(s,1h),7.55(s,1h),7.03-6.51(m,3h),4.94-4.60(m,1h),3.97-3.39(m,13h),3.35-2.19(m,8h),1.75-1.50(m,3h).实施例146,7-二氯-2-(2-(4-(4-氟-2-甲氧基苯基)哌嗪-1-基)乙基)-1-甲基-1,2,3,4-四氢异喹啉(iv-14)二盐酸盐的制备取化合物1-(4-氟-2-甲氧基苯基)哌嗪盐酸盐(0.1mol,1.0eq.)加二氯甲烷150ml溶解,将naoh水溶液(5.2g,50ml)滴加至上述溶液中,室温搅拌0.5h,静置分层,水相30ml二氯甲烷萃取,合并有机相,浓缩得油状物,溶解于30ml丙酮中,加入1-溴-2-氯乙烷(0.11mol,1.1eq.),再加25%naoh水溶液18ml,室温搅拌反应12h停止搅拌,静置分层,水层二氯甲烷萃取,合并有机相,饱和食盐水洗涤,分液,有机相经无水硫酸钠干燥,过滤,浓缩,硅胶柱层析纯化(洗脱剂:二氯甲烷),浓缩,得1-(2-氯乙基)-4-(4-氟-2-甲氧基苯基)哌嗪,类白色固体14.7g。esi-ms[m+h]+:m/z273.1取1-(2-氯乙基)-4-(4-氟-2-甲氧基苯基)哌嗪(4.8mmol,1.0eq.),6,7-二氯-1-甲基-1,2,3,4-四氢异喹啉(4.8mmol,1.0eq.),n,n-二异丙基乙胺2.48g(19.2mmol,4.0eq.),碘化钾0.8g(4.8mmol,1.0eq.)溶于30ml乙腈,升温回流,搅拌6.5h。按合成通法进行后处理操作,得白色固体,目标化合物(iv-14)二盐酸盐,收率74%。esi-ms[m+h]+:m/z452.21h-nmr(400mhz,dmso)δ11.33-11.01(m,2h),7.62(s,1h),7.55(s,1h),7.04-6.51(m,3h),4.95-4.65(m,1h),4.03-2.93(m,19h),1.75-1.51(m,3h).实施例15(r)-8-氯-3-(2-(4-(2,3-二氯苯基)哌嗪-1-基)乙基)-1-甲基-2,3,4,5-四氢-1h-苯骈氮杂(iv-15)盐酸盐的制备取1-(2-氯乙基)-4-(2,3-二氯苯基)哌嗪(4.8mmol,1.0eq.),(r)-8-氯-1-甲基-2,3,4,5-四氢-1h-苯骈氮杂(4.8mmol,1.0eq.),n,n-二异丙基乙胺2.48g(19.2mmol,4.0eq.),碘化钾0.8g(4.8mmol,1.0eq.)溶于30ml乙腈,升温回流,搅拌6.5h。按合成通法进行后处理操作,得白色固体,目标化合物(iv-15)盐酸盐,收率72%。esi-ms[m+h]+:m/z452.11h-nmr(400mhz),dmso-d6:δ11.18-10.90(m,1h),7.42-7.30(m,2h),7.30-7.12(m,4h),3.44-2.70(m,19h),1.36(d,3h).实施例16(r)-8-氯-3-(3-(4-(2,3-二氯苯基)哌嗪-1-基)丙基)-1-甲基-2,3,4,5-四氢-1h-苯骈氮杂(iv-16)二盐酸盐的制备取1-(3-氯丙基)-4-(2,3-二氯苯基)哌嗪(4.8mmol,1.0eq.),(r)-8-氯-1-甲基-2,3,4,5-四氢-1h-苯骈氮杂(4.8mmol,1.0eq.),n,n-二异丙基乙胺2.48g(19.2mmol,4.0eq.),碘化钾0.8g(4.8mmol,1.0eq.)溶于30ml乙腈,升温回流,搅拌6.5h。按合成通法进行后处理操作,得白色固体,目标化合物(iv-16)二盐酸盐,收率75%。esi-ms[m+h]+:m/z466.21h-nmr(400mhz),dmso-d6:δ11.20-10.88(m,2h),7.39(dd,1h),7.37(t,1h),7.34-7.24(m,3h),7.23(dd,1h),3.77-3.55(m,4h),3.50-3.40(m,4h),3.30-3.15(m,8h),3.06-2.96(m,1h),2.94-2.87(m,2h),2.31-2.20(m,2h),1.40(d,3h).实施例172-(3-(4-(2,3-二氯苯基)哌嗪-1-基)丙基)-1-甲基-1,2,3,4-四氢异喹啉-7-甲腈(iv-17)二盐酸盐的制备取1-(3-氯丙基)-4-(2,3-二氯苯基)哌嗪(4.8mmol,1.0eq.),1-甲基-1,2,3,4-四氢异喹啉-7-甲腈(4.8mmol,1.0eq.),n,n-二异丙基乙胺2.48g(19.2mmol,4.0eq.),碘化钾0.8g(4.8mmol,1.0eq.)溶于30ml乙腈,升温回流,搅拌6.5h。按合成通法进行后处理操作,得白色固体,目标化合物(iv-17)二盐酸盐,收率79%。esi-ms[m+h]+:m/z443.21h-nmr(400mhz,dmso)δ11.43-11.20(m,2h),8.01-7.35(m,5h),7.23(dd,1h),4.94-4.60(m,1h),3.97-3.39(m,10h),3.35-2.19(m,8h),1.75-1.50(m,3h).实施例182-(2-(4-(2,3-二氯苯基)哌嗪-1-基)乙基)-1-甲基-1,2,3,4-四氢异喹啉-7-甲腈(iv-18)二盐酸盐的制备取1-(2-氯乙基)-4-(2,3-二氯苯基)哌嗪(4.8mmol,1.0eq.),1-甲基-1,2,3,4-四氢异喹啉-7-甲腈(4.8mmol,1.0eq.),n,n-二异丙基乙胺2.48g(19.2mmol,4.0eq.),碘化钾0.8g(4.8mmol,1.0eq.)溶于30ml乙腈,升温回流,搅拌6.5h。按合成通法进行后处理操作,得白色固体,目标化合物(iv-18)二盐酸盐,收率81%。esi-ms[m+h]+:m/z429.21h-nmr(400mhz,dmso)δ11.40-11.01(m,2h),8.04-7.33(m,5h),7.23(dd,1h),4.92-4.63(m,1h),4.01-2.84(m,16h),1.76-1.50(m,3h).实施例192-(2-(4-(2,3-二氯苯基)哌嗪-1-基)乙基)-1-甲基-1,2,3,4-四氢异喹啉(iv-19)二盐酸盐的制备取1-(3-氯丙基)-4-(2,3-二氯苯基)哌嗪(4.8mmol,1.0eq.),1-甲基-1,2,3,4-四氢异喹啉(4.8mmol,1.0eq.),n,n-二异丙基乙胺2.48g(19.2mmol,4.0eq.),碘化钾0.8g(4.8mmol,1.0eq.)溶于30ml乙腈,升温回流,搅拌6.5h。按合成通法进行后处理操作,得白色固体,目标化合物(iv-19)二盐酸盐,收率77%。esi-ms[m+h]+:m/z404.21h-nmr(400mhz),dmso-d6:δ11.65-11.14(m,2h),7.39(dd,1h),7.36(t,1h),7.33-7.20(dd,5h),4.95-4.68(m,1h),3.92-3.01(m,16h),1.76-1.50(m,3h)实施例202-(2-(4-(2-甲氧基苯基)哌嗪-1-基)乙基)-1-甲基-1,2,3,4-四氢异喹啉(iv-20)二盐酸盐的制备取1-(2-氯乙基)-4-(2-甲氧基苯基)哌嗪(4.8mmol,1.0eq.),1-甲基-1,2,3,4-四氢异喹啉(4.8mmol,1.0eq.),n,n-二异丙基乙胺2.48g(19.2mmol,4.0eq.),碘化钾0.8g(4.8mmol,1.0eq.)溶于30ml乙腈,升温回流,搅拌6.5h。按合成通法进行后处理操作,得白色固体,目标化合物(iv-20)二盐酸盐,收率81%。esi-ms[m+h]+:m/z366.21h-nmr(400mhz),dmso-d6:δ11.35-11.28(m,2h),7.34-7.22(m,4h),7.10-7.68(m,4h),4.95-4.66(m,1h),3.80(s,3h),3.78--3.50(m,8h),3.34-2.05(m,8h),1.74-1.51(m,3h)实施例21制备方法:将活性成分与蔗糖、玉米淀粉混合,加水湿润,搅拌均匀,干燥,粉碎过筛,加入硬脂酸镁,混合均匀,压片。每片重250mg,活性成分含量为25mg。实施例22针剂:本发明的衍生物10mg注射用水90mg制备方法:将活性成分溶解于注射用水,混合均匀,过滤,将所获得的溶液在无菌条件下分装于安瓿瓶中,每瓶10mg,活性成分含量为1mg/瓶。实施例23化合物iv-1~iv-20体外与5-ht1a受体结合试验1实验材料1.1溶媒表1.溶媒相关信息1.2标记配体表2.标记配体相关信息及配制1.3非标记配体表3.非标化合物相关信息1.4cho-5-ht1a细胞:转染人源5-ht1a受体并稳定表达,购于perkinelmer公司。1.5主要仪器表4.仪器信息名称型号生产厂家ph计phs-3c上海精科精密电子天平ms105dumettlertoledo液体闪烁计数仪425-304芬兰hidex液闪仪microbeta2450perkinelmer公司高速离心机j26xpithermo高速分散机ika-t10ika1.6主要试剂及耗材表5.主要试剂及耗材2实验方法2.1配制溶液:50mmtris-hcl缓冲液配制:取96.8gtris溶于双蒸水中总体积为4000ml,用hcl调ph为7.3,稀释到16000ml,ph=7.4。5-ht1a缓冲液配制:称取0.117gedta,1.56mg优降宁,0.4g抗坏血酸,0.48gmgso4,加入50mm的tris-hcl缓冲液总体积为400ml,调整ph=7.4。放射性配体的配制:取放射性配体母液50μl,用无水乙醇稀释成5ml,备用。非标记配体的配制:取5-ht适量,分别用超纯水配成终浓度为2*10-5m的溶液。配制各种受试药物溶液:取各药物适量,先加用dmso稀释到1ml,配成贮备液。临用时再从贮备液中取10μl,用超纯水依次稀释,使其浓度为2*10-5m。甲苯闪烁液:取5.0gppo,0.1gpopop,加入到1000ml甲苯中。2.2制备膜受体:cho-5-ht1acell由-80℃冰箱取出后自然解冻,在1000g,4℃下离心10分钟。取沉淀,弃上清液。沉淀加a液(50mm的tris-hcl缓冲液,1mmedta含0.1%的抗坏血酸、20μm优降宁和10mmmgso4,ph=7.4)。细胞混匀20-30秒,然后50000g,4℃离心15min。小心的弃去上层液,再次加入a液(50mm的tris-hcl缓冲液,1mmedta含0.1%的抗坏血酸、20μm优降宁和10mmmgso4,ph=7.4),混匀,50000g,4℃离心15min离心。重复三次。-80℃储存。2.3受体竞争结合实验:第一步:先将制备好的膜用匀浆液a制成8mg/ml膜的混悬液,备用。第二步:各反应管分别加入膜制备物100μl。第三步:总结合管(tb)加入100μla液,非特异性结合管(nb)加入5-ht100μl(终浓度1.0*10-5m),各受试化合物管(cb)加入100μl受试化合物。第四步:各反应管分别加入放射性配体0.56nm[3h]-8-oh-dpat10μl。第五步:将各反应管25℃温孵60min,反应完毕,结合的配基通过减压快速过滤,whatman试纸gf/c板提前用0.5%pei浸泡1h以上,过滤后将滤膜于烘箱60℃烘干,贴上底膜后加入45μl闪烁液,封好上膜,静置。第六步:将闪烁瓶放入液闪计数仪计数。2.4数据处理和统计分析graphpad计算各化合物ic50;通过scatchard作图得出各放射性配基kd值及bmax;其中:tb代表总结合管cpm值;cb代表待测化合物的cpm值;nsb代表非标的cpm值;l代表放射性配基浓度,单位为nm;kd代表仅在非标存在的情况下放射性配基平衡解离常数,单位为nm;ic50代表待测化合物的半数抑制浓度,单位为nm;3.实验结果化合物体外与5-ht1a受体结合试验见表6。表6.化合物iv-1~iv-20的二盐酸盐与5-ht1a受体亲和试验结果化合物iv-1~iv-6的盐酸盐进行浓度梯度实验,测定与5-ht1a受体亲和作用的ki值,结果如下表7.。表7.化合物的5-ht1a结合作用的ki值试验表明,浓度为10μmol/l时,化合物iv-1~iv-20的盐酸盐具有5-ht1a受体高亲和活性。特别地,5-ht1a受体亲和活性浓度梯度实验结果表明,化合物iv-3~iv-6的二盐酸盐对5-ht1a亲和活性高于阳性药利培酮。实施例24化合物体外与sigma-1受体结合作用试验1实验材料1.1溶媒表8.溶媒相关信息溶媒名称来源批号备注dmso国药集团20150508分析纯超纯水江苏恩华--hcl上海苏懿20150414分析纯冰醋酸上海凌峰20140730分析纯1.2标记配体表9.标记配体相关信息及配制1.3非标记配体及阳性药信息表10.非标及阳性药相关信息1.4清洁级豚鼠,雌雄兼有,300-400g,购于南京青龙山动物养殖中心。1.5主要仪器表11.仪器信息名称型号生产厂家ph计phs-3c上海精科精密电子天平ms105dumettlertoledo液体闪烁计数仪425-304芬兰hidex液闪仪microbeta2450perkinelmer公司高速离心机j26xpithermo高速分散机ika-t10ika1.6主要试剂及耗材表12.主要试剂及耗材名称货号/批号生产厂家无水乙醇20150422永华科技trist1378-1kg/bcbq2051vsigma蔗糖1509221西陇化工microscintps闪烁液94-1249cperkinelmer(赠送)甲苯20151016上海苏懿ppod4630-100g/slbj0333vsigmapopopp3754-5g/031m1737vsigmapei05019jevsigmagf/c滤膜1822-047whatmanunifilter-96gf/c板6005174perkinelmer2实验方法2.1配制溶液:50mmtris-hcl缓冲液配制:取96.8gtris溶于双蒸水中总体积为4000ml,用hcl调ph为7.3,稀释到16000ml,ph=7.4。sigma-1受体缓冲液配制(a):①、0.01m的tris-hcl缓冲液、含0.32m蔗糖溶液,ph=7.4②、0.01m的tris-hcl缓冲液,ph=7.4。放射性配体的配制:取放射性配体母液15μl,用无水乙醇稀释成5ml,备用。非标记配体的配制:取haloperidol适量,分别用超纯水配成终浓度为2*10-5m的溶液。配制各种受试药物溶液:取各药物适量,先加用dmso稀释到1ml,配成贮备液。临用时再从贮备液中取10μl,用超纯水依次稀释,使其浓度为2*10-5m。甲苯闪烁液:取5.0gppo,0.1gpopop,加入到1000ml甲苯中。2.2制备膜受体:豚鼠断头,冰上操作,迅速取脑,加入①匀浆液匀浆,在1000g4℃,离心10min,取上清弃沉淀。然后上清液加于50000g,4℃离心15min;取沉淀加②匀浆液,25℃孵育15min,然后于50000g,4℃离心15min;重复离心。将沉淀于-80℃储存备用。2.3受体竞争结合实验:第一步:先将制备好的膜用匀浆液a制成150mg/ml膜的混悬液,备用。第二步:各反应管分别加入膜制备物100μl。第三步:总结合管(tb)加入100μla液,非特异性结合管(nb)加入haloperidol100μl(终浓度1.0*10-5m),各受试化合物管(cb)加入100μl受试化合物。第四步:各反应管分别加入放射性配体[3h]-pentazocine10μl,终浓度为5nm。第五步:将各反应管25℃温孵135min,反应完毕,结合的配基通过减压快速过滤,96孔抽滤gf/c板提前用0.5%pei浸泡1h以上,过滤后将滤膜于烘箱60℃烘干,贴上底膜后加入45μl闪烁液,封好上膜,静置。第六步:将闪烁瓶放入液闪计数仪计数。2.4数据处理和统计分析graphpad计算各化合物ic50;通过scatchard作图得出各放射性配基kd值及bmax;其中:tb代表总结合管cpm值;cb代表待测化合物的cpm值;nsb代表非标的cpm值;l代表放射性配基浓度,单位为nm;kd代表仅在非标存在的情况下放射性配基平衡解离常数,单位为nm;ic50代表待测化合物的半数抑制浓度,单位为nm。3.实验结果化合物体外与sigma-1受体结合试验见表13.。表13.化合物iv-1~iv-6与sigma-1受体亲和试验结果化合物iv-1~iv-6的盐酸盐进行sigma-1受体结合浓度梯度实验,测定其ki值,结果如下表14.。表14.化合物iv-1~iv-6对sigma-1受体结合作用的ki值试验表明:化合物iv-2、iv-4、iv-5的二盐酸盐具有sigma-1受体高亲和活性,其作用强度与阳性对照药s1ra(lan20130519cas:878141-96-9)相当。化合物iv-3和iv-6具有sigma-1受体较高亲和作用强度。实施例256个化合物的多靶点体外亲和试验1.化合物(cas:1243651-06-0)的制备参照chemmedchem2010,5,1300~1317中的方法制备化合物2-[3-[4-(2,3-二氯苯基)-1-哌嗪基]丙基]-3,4-二氢-1(2h)-异喹啉酮盐酸盐。esi-ms[m+h]+:m/z418.12.化合物(cas:349672-17-9)的制备将1,2,3,4-四氢异喹啉(0.67g,0.005mol),3-溴丙酰氯(0.86g,0.005mol),n,n-二异丙基乙胺(1.94g,0.015mol),20ml二氯甲烷加入到三口烧瓶中,室温下搅拌反应8小时,停止搅拌,静置分层,水层二氯甲烷萃取,合并有机相,饱和食盐水洗涤,分液,有机相经无水硫酸钠干燥,过滤,浓缩得中间体3-溴-1-(3,4-二氢异喹啉-2(1h)-基)丙-1-酮。取3-溴-1-(3,4-二氢异喹啉-2(1h)-基)丙-1-酮(0.54g,0.002mol),2,3-二氯苯基哌嗪(0.46g,0.002mol),n,n-二异丙基乙胺(0.78g,0.006mol),20ml乙腈加入到三口烧瓶中,搅拌下,升温至回流反应6小时。停止加热,减压蒸馏除去溶剂,加入水、乙酸乙酯各100ml,静置分层,水层经乙酸乙酯萃取3次,合并有机相,饱和食盐水洗涤,无水硫酸钠干燥,浓缩后经硅胶柱层析纯化得3-(4-(2,3-二氯苯基)哌嗪-1-基)-1-(3,4-二氢异喹啉-2(1h)-基)丙-1-酮(cas:349672-17-9),白色固体。esi-ms[m+h]+:m/z418.13.化合物(cas:349671-99-4)的制备将1-(3-氯丙基)-4-(2,3-二氯苯基)哌嗪(1.54g,5mmol),吲哚啉(0.6g,5mmol),n,n-二异丙基乙胺(2.58g,20mmol),碘化钾(0.83g,5mmol)溶于30ml乙腈,升温回流,搅拌6h。停止加热,减压蒸馏除去溶剂,加入水、乙酸乙酯各100ml,静置分层,水层经乙酸乙酯萃取3次,合并有机相,饱和食盐水洗涤,无水硫酸钠干燥,浓缩后经硅胶柱层析纯化得游离碱,加30ml乙酸乙酯溶解,滴加3mhcl/etoac调节ph=2~3,固体析出,过滤,真空干燥,得1-(3-(4-(2,3-二氯苯基)哌嗪-1-基)丙基)吲哚啉盐酸盐(cas:349671-99-4)1.3g,白色固体。esi-ms[m+h]+:m/z390.14.化合物体外多靶点结合试验采用同位素配基结合试验,分别进行化合物iv-2、iv-3、iv-4、iv-5、iv-6、cas:349672-17-9、cas:1243651-06-0和cas:349671-99-4进行d1受体、d2受体、d3受体、d4受体、α型肾上腺素能受体α1和α2、sigma-1、sigma-2受体亲和试验测试,具体试验如下。4.1.实验材料4.1.1溶媒表8.溶媒相关信息溶媒名称来源批号备注dmso国药集团20150508分析纯超纯水江苏恩华--hcl上海苏懿20150414分析纯冰醋酸上海凌峰20140730分析纯4.1.2标记配体表9.标记配体相关信息4.1.3非标记配体表10.非标化合物相关信息4.1.4受体膜来源表11.受体膜来源相关信息受体受体膜来源取材部位d1人cho-d1细胞d3人cho-d3细胞d4人cho-d4细胞d2l人cho-d2l细胞α1,α2大鼠脑皮层sigma-1,sigma-2豚鼠豚鼠全脑4.1.5主要仪器表12.仪器信息4.1.6主要试剂及耗材表13.主要试剂及耗材4.1.7对照品相关信息表14.阳性对照品相关信息表对照品名称纯度理化性质保存条件备注risperidone≥99%白色粉末阴凉、干燥室温-duloxetine≥99%白色粉末阴凉、干燥室温-clozapine≥99%白色粉末阴凉、干燥室温-olanzapine≥99%白色粉末阴凉、干燥室温-a-960656≥99%白色粉末阴凉、干燥室温-pcp≥99%白色粉末阴凉、干燥室温-4.2供试品的配制依据文献及本实验室经验,选定1.0*10-5m-1.0*10-10m(终浓度)为实验研究剂量,根据实验结果,对于ki值超出该剂量范围的扩大范围验证。4.3实验方法及步骤4.3.1实验动物信息种属和品系:sd大鼠等级:spf数量和性别:雄42只,雌4只供应单位:浙江省实验动物中心动物生产许可证:scxk(浙)2014-0001动物使用许可证:syxk(苏)2014-00494.3.2组织取材大鼠断头后,打开颅骨,冰上操作分离皮层、纹状体,分离后-80℃冰箱单独存放以供受体实验使用。存放时间不超过6个月。4.3.3匀浆液配制a:(用于制备cho-d1受体膜):称取1.753gnacl,93.1mgkcl,508mgmgcl2.6h2o,55.4mgcacl2,加入50mm的tris-hcl缓冲液总体积为250ml,调整ph=7.4。使其终浓度分别为nacl120mm、kcl5mm、mgcl210mm、cacl22mm。b:(用于制备cho-d2受体膜):称取1.191ghepes,101.6mgmgcl2.6h2o,加入纯化水总体积为250ml,调整ph=7.4。使其终浓度分别为hepes20mm、mgcl22mm。c:(用于制备cho-d3受体膜):称取50.8mgmgcl2.6h2o,加入50mm的tris-hcl缓冲液总体积为250ml,调整ph=7.4。使其终浓度为mgcl21mm。d:(用于制备cho-d4受体膜):称取50.8mgmgcl2.6h2o,加入50mm的tris-hcl缓冲液总体积为250ml,调整ph=7.4。使其终浓度为mgcl21mm。e:(用于制备α1和α2受体膜):称取146mgedta,加入50mm的tris-hcl缓冲液总体积为1000ml,调整ph=7.7。使其终浓度为edta0.5mm。f:(用于制备sigma-1、sigma-2受体膜):①、0.01m的tris-hcl缓冲液、含0.32m蔗糖溶液,ph=7.4.②、0.01m的tris-hcl缓冲液,ph=7.44.3.4受体膜的制备1)cho-d1受体膜的制备cellcho-d1由-80℃冰箱取出后自然解冻,3000g离心5min,沉淀加入匀浆液a,用旋涡混合器混匀,在50000g,4℃离心12min,弃上清液,取沉淀,再次加入a缓冲液(ph=7.4)洗涤,重复三次离心,离心完毕,弃上清液,将沉淀于-80℃储存备用。2)cho-d2受体膜的制备cellcho-d2由-80℃冰箱取出后自然解冻,3000g离心5min,沉淀加入匀浆液b,用旋涡混合器混匀,在50000g,4℃离心12min,弃上清液,取沉淀,再次加入b缓冲液(ph=7.4)洗涤,重复三次离心,离心完毕,弃上清液,将沉淀于-80℃储存备用。3)cho-d3受体膜的制备cellcho-d3由-80℃冰箱取出后自然解冻,2000g离心10min,沉淀加入匀浆液c,用旋涡混合器混匀,在45000g,4℃离心15min,弃上清液,取沉淀,再次加入c缓冲液(ph=7.4)洗涤,重复三次离心,离心完毕,弃上清液,将沉淀于-80℃储存备用。4)cho-d4受体膜的制备cellcho-d4由-80℃冰箱取出后自然解冻,2000g离心10min,沉淀加入匀浆液d,用旋涡混合器混匀,在45000g,4℃离心15min,弃上清液,取沉淀,再次加入d缓冲液(ph=7.4)洗涤,重复三次离心,离心完毕,弃上清液,将沉淀于-80℃储存备用。5)α1和α2(组织)受体膜的制备-80℃冰箱取出大鼠皮层自然解冻,加入匀浆液e,用旋涡混合器混匀,在48000g,4℃离心15min,弃上清液,取沉淀,加入50mm的tris-hcl缓冲液(ph=7.7)洗涤,重复三次离心,离心完毕,弃上清液,将沉淀于-80℃储存备用。6)sigma-1,sigma-2(组织)受体膜的制备豚鼠断头,冰上操作,迅速取脑,加入f匀浆液匀浆,在1000g4℃,离心10min,取上清弃沉淀。然后上清液加于50000g,4℃离心15min;取沉淀加g②匀浆液,25℃孵育15min,然后于50000g,4℃离心15min;重复离心。将沉淀于-80℃储存备用。4.3.5受体竞争结合试验1)cho-d1受体竞争结合试验第一步:先将制备好的膜用匀浆液h制成8mg/ml膜的混悬液备用,备用。第二步:各反应管分别加入膜制备物100μl。第三步:总结合管(tb)加入100μlh液,非特异性结合管(nb)加入butaclamol(终浓度1.0*10-5m)100μl,各受试化合物结合管(cb)加入100μl受试化合物。第四步:各反应管分别加入放射性配体2nm[3h]-sch2339010μl。第五步:将各反应管37℃温孵15min,反应完毕,结合的配基通过减压快速过滤,whatman试纸gf/c提前1h使用0.5%pei溶液饱和,用冰冷的tris缓冲液充分洗涤。过滤后将滤膜60℃烘干,贴上底膜后加入45μl闪烁液,封好上膜,静置。第六步:将闪烁瓶放入液闪计数仪计数。2)cho-d2l受体竞争结合试验第一步:先将制备好的膜用匀浆液i制成8mg/ml膜的混悬液备用,备用。第二步:各反应管分别加入膜制备物100μl。第三步:总结合管(tb)加入100μli液,非特异性结合管(nb)加入100μlhaloperidol(终浓度1.0*10-5m),各受试化合物结合管(cb)加入100μl受试化合物。第四步:各反应管分别加入放射性配体1.176nm[3h]-spiperone10μl。第五步:将各反应管37℃温孵25min,反应完毕,结合的配基通过减压快速过滤whatman试纸gf/b板提前用0.5%pei浸泡1h以上,过滤后将滤膜60℃烘干,贴上底膜后加入45μl闪烁液,封好上膜,静置。第六步:将闪烁瓶放入液闪计数仪计数。3)d3受体竞争结合试验第一步:先将制备好的膜用匀浆液j制成8mg/ml膜的混悬液备用,备用。第二步:各反应管分别加入膜制备物100μl。第三步:总结合管(tb)加入100μlj液,非特异性结合管(nb)加入10μmhaloperidol(终浓度1.0*10-5m)100μl,各受试化合物结合管(cb)加入100μl受试化合物。第四步:各反应管分别加入放射性配体1.176nm[3h]-spiperone10μl。第五步:将各反应管27℃温孵30min,反应完毕,结合的配基通过减压快速过滤whatman试纸gf/c提前1h使用0.5%pei溶液饱和,用冰冷的tris缓冲液充分洗涤,过滤后将滤膜60℃烘干,贴上底膜后加入45μl闪烁液,封好上膜,静置。第六步:将闪烁瓶放入液闪计数仪计数。4)d4受体竞争结合试验第一步:先将制备好的膜用匀浆液j制成8mg/ml膜的混悬液备用,备用。第二步:各反应管分别加入膜制备物100μl。第三步:总结合管(tb)加入100μlj液,非特异性结合管(nb)加入10μmhaloperidol(终浓度1.0*10-5m)100μl,各受试化合物结合管(cb)加入100μl受试化合物。第四步:各反应管分别加入放射性配体1.176nm[3h]-spiperone10μl。第五步:将各反应管27℃温孵30min,反应完毕,结合的配基通过减压快速过滤whatman试纸gf/c提前1h使用0.5%pei溶液饱和,用冰冷的tris缓冲液充分洗涤,过滤后将滤膜60℃烘干,贴上底膜后加入45μl闪烁液,封好上膜,静置。第六步:将闪烁瓶放入液闪计数仪计数。5)α1去甲肾上腺素受体竞争结合试验第一步:先将制备好的膜用匀浆液k制成200mg/ml膜的混悬液备用,备用。第二步:各反应管分别加入膜制备物100μl。第三步:总结合管(tb)加入100μlk液,非特异性结合管(nb)加入prazosin(终浓度1.0*10-5m)100μl,各受试化合物结合管(cb)加入100μl受试化合物。第四步:各反应管分别加入放射性配体0.7nm[3h]-prazosin10μl。第五步:将各反应管25℃温孵60min,反应完毕,结合的配基通过减压快速过滤,whatman试纸gf/c提前1h使用0.5%pei溶液饱和,用冰冷的tris缓冲液充分洗涤,将滤片取出放到2ml闪烁杯中,加入1ml的甲苯闪烁液并混匀。第六步:将闪烁瓶放入液闪计数仪计数。6)α2去甲肾上腺素受体竞争结合试验第一步:先将制备好的膜用匀浆液k制成200mg/ml膜的混悬液备用,备用。第二步:各反应管分别加入膜制备物100μl。第三步:总结合管(tb)加入100μlk液,非特异性结合管(nb)加入rauwolscine(终浓度1.0*10-5m)100μl,各受试化合物结合管(cb)加入100μl受试化合物。第四步:各反应管分别加入放射性配体[3h]-rauwolscine(终浓度1nm)10μl。第五步:将各反应管25℃温孵60min,反应完毕,结合的配基通过减压快速过滤,whatman试纸gf/c提前1h使用0.5%pei溶液饱和,用冰冷的tris缓冲液充分洗涤,将滤片取出放到2ml闪烁杯中,加入1ml的甲苯闪烁液并混匀。第六步:将闪烁瓶放入液闪计数仪计数。7)sigma-1受体竞争结合试验第一步:先将制备好的膜用匀浆液f制成150mg/ml膜的混悬液,备用。第二步:各反应管分别加入膜制备物100μl。第三步:总结合管(tb)加入100μla液,非特异性结合管(nb)加入haloperidol100μl(终浓度1.0*10-5m),各受试化合物管(cb)加入100μl受试化合物。第四步:各反应管分别加入放射性配体[3h]-pentazocine10μl,终浓度为5nm。第五步:将各反应管25℃温孵135min,反应完毕,结合的配基通过减压快速过滤,96孔抽滤gf/c板提前用0.5%pei浸泡1h以上,过滤后将滤膜于烘箱60℃烘干,贴上底膜后加入45μl闪烁液,封好上膜,静置。第六步:将闪烁瓶放入液闪计数仪计数。8)sigma-2受体竞争结合试验第一步:先将制备好的膜用适量的匀浆液f(50mmtris-hcl,ph=7.4),用匀浆分散均匀,调整为0.1g/ml,备用。第二步:各反应孔分别加入膜制备物100μl。第三步:总结合孔(tb)加入100μlf液,非特异性结合孔(nb)加入5μmdtg100μl(终浓度0.5*10-5m),各受试化合物特异性结合孔(sb)加入100μl受试化合物(终浓度1.0*10-5m),再加入100nm(+)--nanm10μl屏蔽sigma-1受体。第四步:各反应孔分别加入放射性配体[3h]-dtg10μl(终浓度5nm)(各反应孔均设2个平行管,加样时各管置于冰上)。第五步:将反应板于25℃温孵135min,反应完毕,结合的配基通过减压快速滤,whatman试纸gf/c提前2h使用0.5%pei溶液饱和,用冰冷的试验缓冲液充分洗涤,过滤后将滤膜于60℃烘30min,贴底膜后加入45μl闪烁液,封好上膜,静置。第六步:将闪烁瓶放入液闪计数仪计数。4.4数据处理和统计分析logit法计算各化合物ic50;通过scatchard作图得出各放射性配基kd值及bmax;其中:tb代表总结合管cpm值;cb代表待测化合物的cpm值;nsb代表非标的cpm值;l代表放射性配基浓度,单位为nm;kd代表仅在非标存在的情况下放射性配基平衡解离常数,单位为nm;ic50代表待测化合物的半数抑制浓度,单位为nm。4.5实验结果6个化合物体外与对d1受体、d2受体、d3受体、d4受体、α型肾上腺素能受体α1和α2、sigma-1、sigma-2受体亲和试验结果见表15.。表15.6个化合物体外与8个靶点亲和试验结果试验表明:化合物iv-2、iv-4和iv-5体外对sigma-1受体具有高亲和作用,化合物iv-5对sigma-2受体具有中等强度的亲和作用。化合物349671-99-4和349672-17-9对d4受体具有高亲和作用,同时对d3受体具有较高亲和作用,而对sigma-1受体无明显亲和作用。化合物1243651-06-0对d3具有高亲和作用,而对sigma-1受体无明显亲和作用。施例26采用小鼠尾悬挂测试化合物iv-5和iv-6二盐酸盐的体内抗抑郁作用1.实验材料1.1主要试剂受试样品:化合物iv-6二盐酸盐及其适当浓度的溶液。对照药品:盐酸度洛西汀cmc-na:购自上海远宏化工有限公司1.2实验动物km小鼠,雌雄兼有,体重在20-25g/只。2.实验方法(小鼠悬尾实验)实验前几天筛选出体重合格的小鼠并分组,实验时分两天进行,d1将小鼠放到悬尾仪的杠杆上6min,记录后4min的不动时间,筛选出不动时间在60s~180s的小鼠并设立60s~90s、90s~120s、120s~150s、150~180s四个层次然后把各个层次小鼠进行随机分组,每组12只,设立空白对照组、阳性对照组和受试药各给药组。d2,小鼠灌胃给药1h后,将小鼠放到悬尾仪的杠杆上6min,记录后4min的不动时间。(不动标准:所谓不动是指小鼠在悬尾仪的杠杆上静止停止挣扎或呈现荡秋千状态。)3.统计学处理方法求出各组小鼠不动时间的平均值,结果用“均值±标准差”表示,将给药组的结果与对照组进行t检验以评价受试药物是否存在抗抑郁性,以p<0.05为有显著性差异。4.试验结果通过实验观察到,与空白对照组相比,盐酸度洛西汀灌胃给药20mg/kg,小鼠不动时间显著减少。化合物iv-5二盐酸盐在灌胃给药10、30、100mg/kg剂量组中,与空白对照组相比,小鼠不动时间明显减少,具有显著性差异。化合物iv-6二盐酸盐在灌胃给药10、30、100mg/kg剂量组中,与空白对照组相比,小鼠不动时间明显减少,具有显著性差异。具体实验结果如下:表15.化合物iv-5和iv-6二盐酸盐的小鼠尾悬挂试验结果注:*p<0.05,**p<0.01实施例27化合物对小鼠僵住症(catalepsy)实验1.试验目的通过口服灌胃给予iv-5二盐酸盐、iv-6二盐酸盐等化合物,进行化合物进行僵住症试验,评估其在不同剂量下引起锥外体系副作用(eps)的程度。观察化合物的毒性不良反应等。2.供试品、对照品及溶媒表16.供试品、对照品及溶媒相关信息1.实验动物种类及品系:icr小鼠,体重范围:24-30g。动物饲养于含有垫料的小鼠盒内,每笼10只。动物房符合普通级要求,控制温度:20-26℃;湿度:40-70%;照明:明暗各12h;噪音:60db以下。2.实验动物标记识别对小鼠尾部用标记笔进行标记识别,以一个原点表示1,一个线条表示5。3.实验原理及方法实验原理:动物的僵住症(catalepsy)是指一种以肌肉强直和运动不能为特征的状态,是研究锥体外系功能状况常用动物模型。通常认为,僵住症与中枢神经系统中乙酰胆碱(acetylcholine,ach)/多巴胺(dopamine,da)功能的平衡失调有关。分组:空白对照组,模型对照组,阳性对照组,各给药组(每组设3—5个剂量)。每组10只小鼠。剂量设计:药物药效作用ed50起,2-3倍比值设置3-5个剂量组。实验方法及步骤:一般症状观察:实验期间观察记录动物行为活动、外观体征等,一般在给药后20min开始观察。试验试验步骤:小鼠按体重分层后,随机分为阴性对照组、空白组、阳性药物各剂量组以及化合物各剂量组,每组10只。空白组灌胃给予相应溶剂50%peg,灌胃体积为0.1ml/10g。灌胃给药30min、60min、90min时,将小鼠两只前爪轻柔地放在长20cm,直径0.3cm,高于工作台5.5cm的小棒上,再将动物后肢轻放于盒底面,记录小鼠两只前爪在棒上保持姿势的持续时间,以30s僵直不动为阳性反应。如果小鼠前爪一直没有放下,60s时终止观察。统计每个化合物剂量组阳性反应动物数。4.主要实验器材抓棒器材:小鼠盒内放置直径0.3cm,高于工作台5cm的不锈钢棒。5.数据统计处理本文各组数据均以表示,并采用spss17.0统计软件进行统计分析。用概率单位法计算ed50。6.结果与讨论表17.iii-3等4个化合物引起小鼠僵住症的ed50值试验及动物观察结果如下:1)阳性药阿立哌唑引起小鼠僵住症的ed50值为2.62mg/kg。2)化合物349671-99-4引起小鼠僵住症的剂量较低,ed50值为12.34mg/kg,观察到与剂量相关的毒性反应为镇静。3)化合物349672-17-9引起小鼠僵住症的剂量较低,ed50值为12.07mg/kg,观察到与剂量相关的毒性反应为镇静、身凉、肌松。4)化合物1243651-06-0引起小鼠僵住症的剂量低,ed50值为3.08mg/kg,观察到与剂量相关的毒性反应为嗜睡、身凉、肌松。5)化合物iv-5二盐酸盐、iv-6二盐酸盐在灌胃给药最大剂量100mg/kg时,未见明显僵住症状,未观察到明显毒性反应。研究结果提示:化合物iv-5二盐酸盐、iv-6二盐酸盐的锥体外系、镇静、嗜睡等副作用小于化合物349671-99-4、349672-17-9和1243651-06-0。实施例28化合物iv-5和iv-6的二盐酸盐的细菌回复突变试验菌种:鼠沙门氏菌组氨酸营养缺陷突变株ta97,ta98,ta100和ta102。结果:实验包括-s9和+s9两个部分,在无s9测试系统中ta98和加s9测试系统中ta975000μg/皿有抑菌作用。其它剂量对所有菌株均无抑菌作用,生长背景良好。所有测试剂量无论在无s9或加s9实验系统中,化合物ⅲ-3和ⅲ-4均未引起任何菌落回变数明显增加,ames试验阴性。上述研究结果表明:化合物iv-5和iv-6体外对5-ht1a受体具有高亲和活性,同时对sigma-1受体具有高亲和活性;化合物iv-5二盐酸盐在小鼠尾悬挂试验中,经灌胃给药,三组剂量下均具有明显抗抑郁作用,其口服吸收良好;化合物iv-6二盐酸盐在小鼠尾悬挂试验中,经灌胃给药,三组剂量下均具有明显抗抑郁作用,其口服吸收良好;化合物iv-5和iv-6的二盐酸盐ames试验均呈阴性。化合物iv-5和iv-6可作为新型抗抑郁新药开发。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1
- Virosaine A的哌嗪基和咪唑基衍生物的组合物在抗急性肾衰药物中的应用
- 用于抑制shp2活性的1-哒嗪-/三嗪-3-基-哌(-嗪)/啶/吡咯烷衍生物及其组合物的制作方法
- 用于治疗肾细胞癌(rcc)的6-氧代-1,6-二氢-哒嗪衍生物的制作方法
- 含有哌嗪基的吲哚类衍生物及其制备方法和应用
- Virosaine A的哌嗪基和咪唑基衍生物的组合物在防治肝脏损伤药物中的应用
- Virosaine A的哌嗪基和咪唑基衍生物的组合物在抗红细胞低下性贫血药物中的应用
- Virosaine A的哌嗪基和咪唑基衍生物的组合物在抗骨质疏松药物中的应用
- Virosaine A的哌嗪基和咪唑基衍生物的组合物在抗急性痛风药物中的应用
- Virosaine A的哌嗪基和咪唑基衍生物的组合物在预防或治疗胰腺纤维化药物中的应用
- Virosaine A的哌嗪基和咪唑基衍生物的组合物在抗缺氧药物中的应用