一种1,10‑癸二酸的制备方法与流程
本发明涉及化学物质制备技术领域,尤其涉及一种1,10-癸二酸的制备方法。
背景技术:
1,10-癸二酸用途广泛,主要用来制取癸二酸的酯类,其酯类用途广泛,如癸二酸二丁酯、癸二酸二辛酯、癸二酸二异辛酯。这些酯类可作塑料、耐寒橡胶的增塑剂;也可用于制取聚酰胺、聚氨酯、醇酸树脂、合成润滑油、润滑油添加剂以及香料、涂料、化妆品等;也可用作生产尼龙10,10、尼龙9,10、尼龙8,10、尼龙6,10、尼龙9的原料及耐高温润滑油二乙基己酯的原料;也是生产醇酸树脂(用作表面涂料、增塑硝酸纤维素涂料和尿素树脂清漆)和聚氨基甲酸酯橡胶、纤维素树脂、乙烯基树脂及合成橡胶的增塑剂、软化剂和溶剂的原料。
1,10-癸二酸最早是用蓖麻油加碱干馏获得,后续发展了合成方法。目前正在开发石油正构烷烃发酵制1,10-癸二酸的新路线。1,10-癸二酸的另一种生产方法是日本旭化成工业公司开发的己二酸电解氧化法,但是上述方法采用的原料或者来源受限,或者采用的是不可再生的石油资源。
技术实现要素:
基于背景技术存在的技术问题,本发明提出了一种1,10-癸二酸的制备方法,本发明原料廉价易得,原子经济性好,产物收率高,产物纯度高,对环境污染小,工艺路线简单,操作方便,适合工业化大规模生产。
本发明提出的一种1,10-癸二酸的制备方法,包括如下步骤:将物质a在酸性环境中水解开环得到4,7-二酮-1,10-癸二酸;4,7-二酮-1,10-癸二酸与氢气、三氟甲磺酸盐、加氢催化剂加氢脱氧得到1,10-癸二酸,其中,物质a的化学结构式如下:
其中,r为氢原子或烷基。
优选地,r为氢原子或碳原子数小于5的烷基。
优选地,酸性环境的ph小于1。
优选地,三氟甲磺酸盐为+3至+6价的三氟甲磺酸金属盐。
优选地,4,7-二酮-1,10-癸二酸、三氟甲磺酸盐的摩尔比为10-100:1。
上述三氟甲磺酸盐包括w(otf)6、hf(otf)4、al(otf)3、ta(otf)5、nb(otf)5、zr(otf)4等+3至+6价的三氟甲磺酸金属盐。
优选地,加氢催化剂为含有第ⅷ族过渡金属元素的催化剂。
优选地,含有第ⅷ族过渡金属元素的催化剂中的金属元素与4,7-二酮-1,10-癸二酸的摩尔比为1:10-1000。
上述加氢催化剂是指化合物与氢加成时使用的催化剂,如:第ⅷ族过渡金属,含有第ⅷ族过渡金属元素的金属氧化物、金属络合物等;金属络合物的载体一般为氧化铝或活性碳;具体加氢催化剂包括:钯碳、铂碳等。
优选地,加氢脱氧的反应溶剂为羧酸。
优选地,加氢脱氧的反应溶剂为乙酸。
优选地,用酸性物质维持酸性环境。
优选地,酸性物质为无机强酸。
上述无机强酸包括盐酸、硫酸等常规无机强酸。
优选地,氢气压力为3-100atm。
优选地,氢气压力为3-50atm。
优选地,加氢脱氧的反应温度为120-250℃。
优选地,加氢脱氧的反应温度为160-200℃。
优选地,加氢脱氧的反应时间为1-24h。
优选地,水解开环的具体步骤为:将甲醇、物质a混匀,调节ph小于1,回流,旋干得到固体即为4,7-二酮-1,10-癸二酸。
优选地,在水解开环的过程中,用酸性物质水溶液调节ph小于1。
优选地,酸性物质水溶液的质量分数为20-50wt%。
优选地,在水解开环的过程中,回流10-24h。
优选地,在水解开环的过程中,旋干后,再纯化得到4,7-二酮-1,10-癸二酸。
优选地,纯化的具体步骤为:将固体溶于碱性水溶液中,加活性碳脱色,过滤取滤液,调节ph小于1,析晶得到4,7-二酮-1,10-癸二酸。
优选地,加氢脱氧的具体步骤为:将4,7-二酮-1,10-癸二酸、加氢催化剂、三氟甲磺酸盐、反应溶剂混匀,在氢气氛围中,搅拌升温,保温搅拌得到1,10-癸二酸。
优选地,在加氢脱氧过程中,保温搅拌后,再纯化得到1,10-癸二酸。
优选地,纯化的具体步骤为:过滤取滤液,减压蒸馏得到固体;将固体脱色得到1,10-癸二酸。
优选地,脱色的具体步骤为:将固体溶于氢氧化钠水溶液中,加入活性炭脱色,过滤取滤液,用盐酸调节ph小于2,过滤取滤饼,烘干得到1,10-癸二酸。
优选地,糠醛和物质b经碱性物质催化缩合得到物质a,其中,物质b的化学结构式如下:
其中,物质b与物质a中的r相同。
用糠醛和物质b经碱性物质催化缩合得到物质a,是常规的aldol缩合反应,其合成路线如下:
aldol缩合反应的制备方法已经相当完善,本领域技术人员可以根据本领域常规方法进行制备,也可以按照如下限定的内容进行制备:
优选地,r为氢原子时,碱性物质为含金属元素的碱性物质。
上述含金属元素的碱性物质包括氢氧化钠、氢氧化钾、碳酸钠、碳酸钾、乙醇钠、叔丁醇钾等常规含金属元素的碱性物质。
优选地,r为氢原子时,碱性物质与物质b的摩尔比大于1。
优选地,r为烷基时,碱性物质为有机碱。
优选地,r为烷基时,碱性物质为有机碱,其中,有机碱中含有伯胺基团、仲胺基团、叔胺基团中的至少一种基团。
上述有机碱包括吡咯、三乙胺、吗啉、吡啶等常规有机碱。
优选地,r为氢原子时,缩合后,需经酸化处理得到物质a。
优选地,r为氢原子时,缩合的具体步骤为:将碱性物质与物质b溶液混匀,加热至回流,滴加糠醛溶液,回流1-24h,酸化得到物质a。
优选地,r为氢原子时,在缩合过程中,物质b溶液的溶剂为水。
优选地,r为氢原子时,在缩合过程中,糠醛溶液的溶剂为甲醇。
优选地,r为氢原子时,在缩合过程中,用酸性水溶液进行酸化。
优选地,酸性水溶液为质量分数为5-10wt%盐酸水溶液。
优选地,r为氢原子时,在缩合过程中,酸化后,再纯化得到物质a。
优选地,纯化的具体步骤为:过滤取滤饼,冰水洗涤,热水重结晶得到物质a。
优选地,r为氢原子时,在缩合过程中,物质b、糠醛的摩尔比为5:1-3。
优选地,r为烷基时,缩合的具体步骤为:将碱性物质、物质b、糠醛混匀,室温搅拌1-24h得到物质a。
优选地,r为烷基时,在缩合过程中,室温搅拌后,再纯化得到物质a。
优选地,纯化的具体步骤为:过滤取滤液,浓缩得到浓缩物;用乙酸乙酯溶解浓缩物,用硅胶柱洗脱,旋干得到物质a。
优选地,r为烷基时,在缩合过程中,碱性物质、物质b、糠醛的摩尔比为1:1-10:1-10。
上述水解开环过程中,不规定甲醇的用量,根据具体操作确定其用量;不规定回流温度,保持回流状态即可。
上述加氢脱氧过程中,不规定反应溶剂的用量,根据具体操作确定其用量。
上述r为氢原子时的缩合过程中,不规定物质a溶液、糠醛溶液的浓度,根据具体操作确定其浓度;不规定回流温度,保持回流状态即可。
上述加氢脱氧、缩合、水解开环过程中,可以用薄层色谱辅助监测反应是否完全。
本发明的合成路线如下:
本发明人经过深入研究发现,物质a(δ-糠基乙酰丙酸或其酯)在酸性环境中水解开环得到4,7-二酮-1,10-癸二酸,4,7-二酮-1,10-癸二酸中的酮羰基被氢化转变为仲醇羟基后,在三氟甲磺酸盐促进下,仲醇羟基容易和羧酸作用得到仲醇酯;而仲醇酯在三氟甲磺酸盐促进下,可以氢解去除羟基,从而使得4,7-二酮-1,10-癸二酸在加氢催化剂和三氟甲磺酸盐共催化体系下选择性加氢脱氧得到1,10-癸二酸;本发明原子经济性好,产物收率高,产物纯度高,对环境污染小,工艺路线简单,操作方便,适合工业化大规模生产,产物1,10-癸二酸的应用领域广泛,市场前景好,产物的附加值高,本发明具有潜在的工业应用前景;且选用糠醛和物质b(乙酰丙酸或其酯)为原料制备物质a,糠醛可利用半纤维素水解得到,物质b可利用纤维素水解或醇解得到,来源广泛,为可再生的生物质资源,廉价易得,操作简单,降低生产成本,适合工业化生产。
附图说明
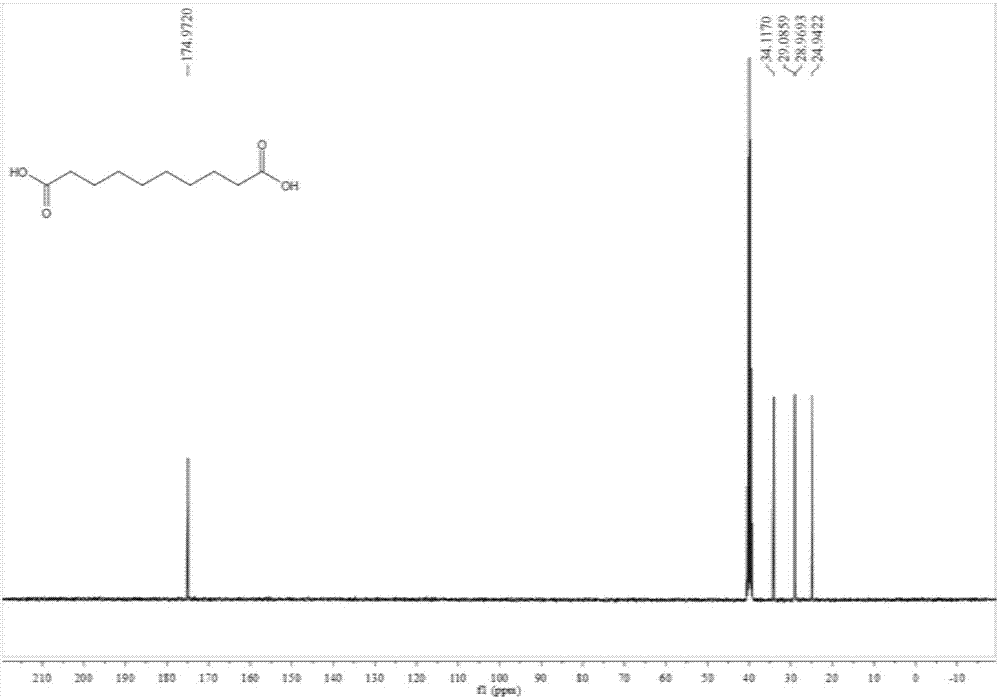
图1为本发明制备得到的1,10-癸二酸的核磁氢谱图。
图2为本发明制备得到的1,10-癸二酸的核磁碳谱图。
图3为本发明制备得到的4,7-二酮-1,10-癸二酸的核磁氢谱图。
图4为本发明制备得到的4,7-二酮-1,10-癸二酸的核磁碳谱图。
具体实施方式
下面,通过具体实施例对本发明的技术方案进行详细说明。
实施例1
一种1,10-癸二酸的制备方法,包括如下步骤:将物质a在酸性环境中水解开环得到4,7-二酮-1,10-癸二酸;4,7-二酮-1,10-癸二酸与氢气、三氟甲磺酸盐、加氢催化剂加氢脱氧得到1,10-癸二酸
实施例2
一种1,10-癸二酸的制备方法,包括如下步骤:将甲醇、δ-糠基乙酰丙酸甲酯混匀,用质量分数为37wt%盐酸水溶液调节ph小于1,回流17h,旋干得到固体;将固体溶于氢氧化钠水溶液中,加活性碳加热脱色,过滤取滤液,用盐酸调节ph小于1,析晶得到4,7-二酮-1,10-癸二酸;
将4,7-二酮-1,10-癸二酸、钯碳、al(otf)3、乙酸混匀,在氢气氛围中,调节氢气压力为80atm,搅拌升温至140℃,保温搅拌20h,过滤取滤液,减压蒸馏得到固体;将固体溶于氢氧化钠水溶液中,加入活性炭脱色,过滤取滤液,用盐酸调节ph小于2,过滤取滤饼,烘干得到1,10-癸二酸,其中,4,7-二酮-1,10-癸二酸、w(otf)6的摩尔比为55:1,钯碳中的钯元素与4,7-二酮-1,10-癸二酸的摩尔比为1:500。
实施例3
一种1,10-癸二酸的制备方法,包括如下步骤:
将氢氧化钠与乙酰丙酸水溶液混匀,加热至回流,滴加糠醛甲醇溶液,回流1h,用质量分数为5wt%盐酸水溶液酸化,过滤取滤饼,冰水洗涤,热水重结晶得到δ-糠基乙酰丙酸,其中,乙酰丙酸、糠醛的摩尔比为5:3,氢氧化钠与乙酰丙酸的摩尔比为1.05:1;
将甲醇、δ-糠基乙酰丙酸混匀,用质量分数为20wt%盐酸水溶液调节ph小于1,回流10h,旋干得到固体;将固体溶于质量分数为10wt%氢氧化钠水溶液中,加活性碳加热脱色,过滤取滤液,用盐酸调节ph小于1,析晶得到4,7-二酮-1,10-癸二酸;
将4,7-二酮-1,10-癸二酸、铂碳、zr(otf)4、乙酸混匀,在氢气氛围中,调节氢气压力为100atm,搅拌升温至120℃,保温搅拌24h,过滤取滤液,减压蒸馏得到固体;将固体溶于氢氧化钠水溶液中,加入活性炭脱色,过滤取滤液,用盐酸调节ph小于2,过滤取滤饼,烘干得到1,10-癸二酸,其中,4,7-二酮-1,10-癸二酸、zr(otf)4的摩尔比为100:1,铂碳中的铂元素与4,7-二酮-1,10-癸二酸的摩尔比为1:10。
实施例4
一种1,10-癸二酸的制备方法,包括如下步骤:
将乙醇钠与乙酰丙酸水溶液混匀,加热至回流,滴加糠醛甲醇溶液,回流24h,用质量分数为10wt%盐酸水溶液酸化,过滤取滤饼,冰水洗涤,热水重结晶得到δ-糠基乙酰丙酸,其中,乙酰丙酸、糠醛的摩尔比为5:1,乙醇钠与乙酰丙酸的摩尔比为1.05:1;
将甲醇、δ-糠基乙酰丙酸混匀,用质量分数为50wt%硫酸水溶液调节ph小于1,回流24h,旋干得到固体;将固体溶于质量分数为10wt%氢氧化钠水溶液中,加活性碳加热脱色,过滤取滤液,用盐酸调节ph小于1,降温析晶得到4,7-二酮-1,10-癸二酸;
将4,7-二酮-1,10-癸二酸、钯碳、hf(otf)4、乙酸混匀,在氢气氛围中,调节氢气压力为3atm,以600r/min的速度,在30min内搅拌升温至250℃,保温搅拌1h,过滤取滤液,减压蒸馏得到固体;将固体溶于氢氧化钠水溶液中,加入活性炭脱色,过滤取滤液,用盐酸调节ph小于2,过滤取滤饼,烘干得到1,10-癸二酸,其中,4,7-二酮-1,10-癸二酸、hf(otf)4的摩尔比为10:1,钯碳中的钯元素与4,7-二酮-1,10-癸二酸的摩尔比为1:1000。
实施例5
一种1,10-癸二酸的制备方法,包括如下步骤:
将三乙胺、乙酰丙酸乙酯、糠醛混匀,室温搅拌1h,过滤取滤液,浓缩得到浓缩物;用乙酸乙酯溶解浓缩物,用硅胶柱洗脱,旋干得到δ-糠基乙酰丙酸乙酯,其中,三乙胺、乙酰丙酸乙酯、糠醛的摩尔比为1:1:1;
将甲醇、δ-糠基乙酰丙酸乙酯混匀,用质量分数为30wt%盐酸水溶液调节ph小于1,回流12h,旋干得到固体;将固体溶于质量分数为10wt%氢氧化钠水溶液中,加活性碳加热脱色,过滤取滤液,用盐酸调节ph小于1,降温至0℃,析晶10h得到4,7-二酮-1,10-癸二酸;
将4,7-二酮-1,10-癸二酸、铂碳、ta(otf)5、乙酸混匀,在氢气氛围中,调节氢气压力为10atm,以700r/min的速度,在30min内搅拌升温至200℃,保温搅拌12h,过滤取滤液,减压蒸馏得到固体;将固体溶于氢氧化钠水溶液中,加入活性炭脱色,过滤取滤液,用盐酸调节ph小于2,过滤取滤饼,烘干得到1,10-癸二酸,其中,4,7-二酮-1,10-癸二酸、ta(otf)5的摩尔比为80:1,铂碳中的铂元素与4,7-二酮-1,10-癸二酸的摩尔比为1:700。
实施例6
一种1,10-癸二酸的制备方法,包括如下步骤:
将吡啶、乙酰丙酸丁酯、糠醛混匀,室温搅拌24h,过滤取滤液,浓缩得到浓缩物;用乙酸乙酯溶解浓缩物,用硅胶柱洗脱,旋干得到δ-糠基乙酰丙酸丁酯,其中,吡啶、乙酰丙酸丁酯、糠醛的摩尔比为1:10:10;
将甲醇、δ-糠基乙酰丙酸丁酯混匀,用质量分数为35wt%硫酸水溶液调节ph小于1,回流20h,旋干得到固体;将固体溶于质量分数为10wt%氢氧化钠水溶液中,加活性碳加热脱色,过滤取滤液,用盐酸调节ph小于1,降温至0℃,析晶10h得到4,7-二酮-1,10-癸二酸;
将4,7-二酮-1,10-癸二酸、钯碳、nb(otf)5、乙酸混匀,在氢气氛围中,调节氢气压力为60atm,以650r/min的速度,在30min内搅拌升温至220℃,保温搅拌8h,过滤取滤液,减压蒸馏得到固体;将固体溶于氢氧化钠水溶液中,加入活性炭脱色,过滤取滤液,用盐酸调节ph小于2,过滤取滤饼,烘干得到1,10-癸二酸,其中,4,7-二酮-1,10-癸二酸、nb(otf)5的摩尔比为40:1,钯碳中的钯元素与4,7-二酮-1,10-癸二酸的摩尔比为1:100。
实施例7
一种1,10-癸二酸的制备方法,包括如下步骤:
将吡咯、乙酰丙酸甲酯、糠醛混匀,室温搅拌8h,过滤取滤液,浓缩得到浓缩物;用乙酸乙酯溶解浓缩物,用硅胶柱洗脱,旋干得到δ-糠基乙酰丙酸甲酯,其中,吡咯、乙酰丙酸甲酯、糠醛的摩尔比为1:8:8;
将甲醇、δ-糠基乙酰丙酸甲酯混匀,用质量分数为40wt%硫酸水溶液调节ph小于1,回流15h,旋干得到固体;将固体溶于质量分数为10wt%氢氧化钠水溶液中,加活性碳加热脱色,过滤取滤液,用盐酸调节ph小于1,降温至0℃,析晶10h得到4,7-二酮-1,10-癸二酸,其中,δ-糠基乙酰丙酸甲酯和硫酸的摩尔比为1:2.8,δ-糠基乙酰丙酸甲酯和甲醇的摩尔体积(mol/l)比为1:1;
将4,7-二酮-1,10-癸二酸、钯含量为5wt%的钯碳、hf(otf)3、乙酸混匀,通氢气置换四次气体后,室温下充氢气至氢气压力为50atm,以800r/min的速度,在30min内搅拌升温至160℃,保温搅拌15h,降温,泄压,过滤取滤液,减压蒸馏得到固体;将固体溶于质量分数为10wt%氢氧化钠水溶液中,加入活性炭加热脱色30min,过滤取滤液,用盐酸调节ph小于2,过滤取滤饼,烘干得到1,10-癸二酸,其中,4,7-二酮-1,10-癸二酸、al(otf)3的摩尔比为50:3,钯碳中的钯元素与4,7-二酮-1,10-癸二酸的摩尔比为1:50,4,7-二酮-1,10-癸二酸和乙酸的摩尔体积(mol/l)比为1:15。
实施例8
一种1,10-癸二酸的制备方法,包括如下步骤:
将碳酸钠分批加入浓度为1.55mol/l乙酰丙酸水溶液中混匀,加热至回流,滴加浓度为5.81mol/l糠醛甲醇溶液,回流1h,用质量分数为7.5wt%盐酸水溶液酸化,过滤取滤饼,冰水洗涤,热水重结晶得到δ-糠基乙酰丙酸,其中,乙酰丙酸、糠醛的摩尔比为1:0.6,碳酸钠与乙酰丙酸的摩尔比为1.46:1;
将甲醇、δ-糠基乙酰丙酸混匀,用质量分数为37wt%盐酸水溶液调节ph小于1,回流15h,旋干得到固体;将固体溶于质量分数为10wt%氢氧化钠水溶液中,加活性碳加热脱色,过滤取滤液,用盐酸调节ph小于1,降温至0℃,析晶10h得到4,7-二酮-1,10-癸二酸,其中,δ-糠基乙酰丙酸和盐酸的摩尔比为1:7.2,δ-糠基乙酰丙酸和甲醇的摩尔体积(mol/l)比为3:4;
将4,7-二酮-1,10-癸二酸、钯含量为10wt%的钯碳、w(otf)6、乙酸混匀,通氢气置换四次气体后,室温下充氢气至氢气压力为30atm,以500r/min的速度,在30min内搅拌升温至180℃,保温搅拌10h,降温,泄压,过滤取滤液,减压蒸馏得到固体;将固体溶于质量分数为10wt%氢氧化钠水溶液中,加入活性炭加热脱色30min,过滤取滤液,用盐酸调节ph小于2,过滤取滤饼,烘干得到1,10-癸二酸,其中,4,7-二酮-1,10-癸二酸、w(otf)6的摩尔比为50:3,钯碳中的钯元素与4,7-二酮-1,10-癸二酸的摩尔比为1:50,4,7-二酮-1,10-癸二酸和乙酸的摩尔体积(mol/l)比为1:10。
对实施例8制备得到的4,7-二酮-1,10-癸二酸、1,10-癸二酸进行核磁检测,结果参照图1-4。
图1为本发明制备得到的1,10-癸二酸的核磁氢谱图,结果为1hnmr(400mhz,dmso):δ=11.98(s,2h),2.18(t,j=7.4hz,4h),1.55-1.40(m,4h),1.25(s,8h);
图2为本发明制备得到的1,10-癸二酸的核磁碳谱图,结果为13cnmr(101mhz,dmso):δ=174.97,34.12,29.03(d,j=11.7hz),24.94;
图3为本发明制备得到的4,7-二酮-1,10-癸二酸的核磁氢谱图,结果为1h-nmr(400mhz,dmso):δ=12.10(s,2h),2.66(dd,j=11.4,4.6hz,8h),2.38(t,j=6.5hz,4h);
图4为本发明制备得到的4,7-二酮-1,10-癸二酸的核磁碳谱图,结果为13c-nmr(101mhz,dmso):δ=208.30,174.17,37.04,36.06,28.12;
由上述检测结果可以看出本发明制备得到是4,7-二酮-1,10-癸二酸、1,10-癸二酸。
统计实施例7、8的收率,检测δ-糠基乙酰丙酸、δ-糠基乙酰丙酸甲酯、4,7-二酮-1,10-癸二酸和1,10-癸二酸的纯度,结果如下:
由上表可以看出本发明的收率高,制备得到的1,10-癸二酸纯度好。
以上所述,仅为本发明较佳的具体实施方式,但本发明的保护范围并不局限于此,任何熟悉本技术领域的技术人员在本发明揭露的技术范围内,根据本发明的技术方案及其发明构思加以等同替换或改变,都应涵盖在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!