一种常温下制备2,4‑二氟苯硼酸的方法与流程
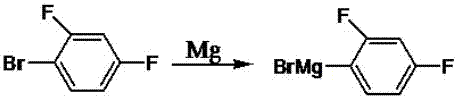
本发明涉及有机合成
技术领域:
,具体涉及一种常温下制备2,4-二氟苯硼酸的方法。
背景技术:
:芳基硼酸性质稳定,在空气中不易被氧化潮解,且与卤代芳烃的suzuki偶联反应有较好的位置及立体选择性,反应条件温和。含氟苯硼酸是芳基硼酸重要代表之一,氟原子具有很强的吸电子,往往会使原来分子的电子性质发生很大的变化,从而使含氟苯硼酸具有良好的稳定性和大的介电各向异性,近年在有机合成、生物医学、农药及材料学方面都有非常重要的作用,在液晶显示材料方面作用尤为显著。2,4-二氟苯硼酸(2,4-diflorophenyl-1-boronicacid)是一种含氟的单取代芳基硼酸化合物,其化学结构式如下:目前合成单取代芳基硼酸方法主要有格氏试剂法、有机锂试剂法、“一锅法”和超声法。格氏试剂法通常需要低温、无水和无氧等条件,反应条件苛刻;有机锂试剂法也是低温操作,耗能大,且有机锂试剂价格昂贵,易爆炸,操作危险性较高;“一锅法”常温下操作,反应条件较简单,仍需严格无水无氧;超声法操作简单,反应速度快,收率高,为单取代芳基硼酸提供了一条简捷的潜在合成途径。专利cn102146099b以2,4-二氟溴苯为原料,在-35℃下将2,4-二氟溴苯制成格氏试剂后,在-60℃下与硼酸酯反应合成2,4-二氟苯硼酸,反应条件苛刻,称重收率比较低,仅为40%。有机锂试剂法(frohnhj.,etal.,zanorgallgchem,2002,2827-2833.)在-70℃下反应,2,4-二氟苯硼酸称重收率为77%,其反应条件苛刻,操作危险性高,且试剂价格昂贵。由于2,4-二氟苯硼酸是氟邻位的单取代芳基硼酸化合物,产生的中间体2,4-二氟苯基溴化镁不稳定易生成副产物,且副产物难以通过重结晶法除去,目前尚未见到“一锅法”或超声法直接合成2,4-二氟苯硼酸的文献报道。针对传统方法合成2,4-二氟苯硼酸过程中,反应温度过低,条件苛刻,操作危险性高,反应收率低及副产物难以分离等不足,本发明公开一种在常温下安全高效合成2,4-二氟苯硼酸的方法,该方法主要通过瞬时控制反应液中2,4-二氟溴苯的浓度,使2,4-二氟苯基溴化镁浓度保持在较低水平,再利用超声辐射加快2,4-二氟苯基溴化镁转化成2,4-二氟苯硼酸,大大减少副产物的产生,并使用萃取-反萃取联合技术除去副产物,不但提高产物收率,同时提高产物的纯度。本发明使用的原料廉价易得,反应条件温和,工艺简单安全,生产成本低,所得产品收率高、纯度好,具有重要的产业化前景。技术实现要素:本发明目的在于提供一种常温下制备2,4-二氟苯硼酸的方法。本发明的技术方案是:1.一种常温下制备2,4-二氟苯硼酸的方法,其特征在于:以硼酸酯、镁屑为底物,在一定超声条件下,用滴加方式加入2,4-二氟溴苯和醚类溶剂的预混液开始合成反应,在反应一定程度后得到2,4-二氟苯硼酸酯反应液,再酸化后得到产品粗液,再通过萃取、反萃取得到高纯度2,4-二氟苯硼酸产品;所述硼酸酯为硼酸三异丙酯、硼酸三乙酯、硼酸三丁酯中的一种;所述醚类溶剂为四氢呋喃、甲基四氢呋喃、乙醚中的一种;所述硼酸酯、镁屑的摩尔比为1:1~6;所述预混液为2,4-二氟溴苯与醚类溶剂组成的混合物,体积比为1:8~16;所述超声条件为超声功率为150w~250w,超声温度0~50℃;所述滴加方式为匀速滴加,滴加时间20min~50min,使2,4-二氟溴苯浓度保持在0.17~0.41mmol/ml;所述反应一定程度为滴加结束后,在超声条件下继续反应60~90min,反应液最终变为淡黄色浊液。2.如权利要求1所述一种常温下制备2,4-二氟苯硼酸的方法,硼酸酯、镁屑、2,4-二氟溴苯摩尔比为1:1~4:1~2。3.如权利要求1所述一种常温下制备2,4-二氟苯硼酸的方法,所述的酸化是将2,4-二氟苯硼酸酯反应液除去未反应的镁屑后,通过滴加0.5mol/l的盐酸调节滤液ph至1~4。4.如权利要求1所述一种常温下制备2,4-二氟苯硼酸的方法,所述的萃取包括:用乙酸乙酯将酸化后的反应液萃取再分液,得到有机相i和水相i,取有机相i再水洗分液得到有机相ii和水相ii,合并水相得到水相iii,再用乙酸乙酯萃取水相ⅲ得到有机相iii,将有机相ii与有机相iii合并得到有机相iv,再在35℃~60℃下旋蒸,除去醚类溶剂和乙酸乙酯,析出的棕黄色固体即2,4-二氟苯硼酸粗产品,最高收率达到73%,纯度80%以上。5.如权利要求1所述一种常温下制备2,4-二氟苯硼酸的方法,所述的反萃取包括:通过向2,4-二氟苯硼酸粗产品中滴入5~10ml0.5mol/l的naoh溶液至ph为10~13,形成2,4-二氟苯硼酸钠溶液,通过乙酸乙酯萃取分液得到有机相i和水相i,取有机相i再用10-20ml纯水洗2-3次得到有机相ii和水相ii,合并水相i和水相ii得到水相iii,再用0.5mol/l的盐酸溶液将水相iii的ph调节到1~3,使2,4-二氟苯硼酸钠酸化形成2,4-二氟苯硼酸混合物,再用乙酸乙酯萃取2-3次,旋蒸除去乙酸乙酯,得到白色高纯度2,4-二氟苯硼酸,两步收率最高达到70%,纯度95~99%。本发明的有益效果:本发明克服了传统方法中反应温度过低、条件苛刻、操作危险性高、反应收率低及副产物难以分离等不足,本方法原料廉价易得,反应条件温和,只需常温下反应,操作简单安全,生产成本低,最高收率达到70%,纯度达到95~98%,具有重要的产业化前景。附图说明图1为合成2,4-二氟苯硼酸的工艺流程图。图2为2,4-二氟苯基溴化镁生成反应式。图3为2,4-二氟苯硼酸生成反应式。图4为主要副反应反应式。图5为2,4-二氟苯硼酸红外图谱3086.39cm-1归属于苯环上的c-h吸收峰,1435.46cm-1、1614.93cm-1归属于苯环骨架变形振动,1400cm-1-1100cm-1为c-f收缩振动吸收,733.20cm-1、682.70cm-1归属于苯环邻位三取代c-h面外弯曲振动吸收,3403.10cm-1归属于缔合的o-h的伸缩振动吸收,1342.25cm-1归属于b-o的伸缩振动吸收。具体实施方式实施例1、“一锅法”合成2,4-二氟苯硼酸在装有冷凝管,恒压滴定漏斗的三口烧瓶中加入0.3mol(0.76g)镁屑、0.1mol(1.88g)硼酸三异丙酯和一颗搅拌子,将0.15mol(2.88g)2,4-二氟溴苯和20ml四氢呋喃加入60ml恒压滴定漏斗,在氮气保护下,边滴加边反应,反应温度40℃,60min滴完,滴完后继续反应180min。反应结束后,滤除未反应的镁屑。向滤液中加入5ml0.1mol/l盐酸,溶液将出现白色絮状物,继续添加5ml0.1mol/l的盐酸,絮状物消失,溶液变澄清,用20ml乙酸乙酯萃取分液,有机相用15ml纯水洗涤一次,水相用乙酸乙酯萃取2×10ml,然后合并有机相,在55℃下旋蒸30min得0.88g粗产物。经液相色谱检测产品纯度为54.46%,产品收率为30.33%。实施例2、超声法合成2,4-二氟苯硼酸在装有冷凝管的100ml圆底烧瓶中加入0.3mol(0.76g)镁屑、0.10mol(1.88g)硼酸三异丙酯,20ml四氢呋喃、0.15mol(2.88g)2,4-二氟溴苯,开启超声反应,超声功率175w,反应温度40℃,反应40min。反应结束后,向滤液中加入5ml0.1mol/l盐酸,溶液将出现白色絮状物,继续添加5ml0.1mol/l的盐酸,絮状物消失,溶液变澄清,用20ml乙酸乙酯萃取分液,有机相用15ml纯水洗涤一次,水相用乙酸乙酯萃取2×10ml,然后合并有机相,在55℃下旋蒸30min得0.97g粗产物。经液相色谱检测产品纯度为67.28%,产品收率为41.31%。实施例3、本发明单因素试验考察取镁屑、硼酸三异丙酯加入圆底烧瓶,取2,4-二氟溴苯、醚类溶剂加入恒压滴定漏斗,边滴加超声。反应结束后,滤除未反应的镁屑,向滤液中加入5ml0.1mol/l盐酸,溶液将出现白色絮状物,继续添加5ml0.1mol/l的盐酸,絮状物消失,溶液变澄清,用20ml乙酸乙酯萃取分液,有机相用15ml纯水洗涤一次,水相用乙酸乙酯萃取2×10ml,然后合并有机相,在60℃下旋蒸得粗产物。用液相色谱检测产品纯度,并计算产品收率。(1)在2,4-二氟溴苯0.15mol(2.88g),硼酸三异丙酯0.10mol(1.88g),反应温度为40℃,反应功率为175w,四氢呋喃用量为20ml,反应时间为100min,滴加时间为30min条件下,镁屑与硼酸三异丙酯的摩尔比影响如表1所示:表1镁:硼酸三异丙酯摩尔比1:12:13:14:1产物质量/g0.961.151.341.38产物纯度/%32.4382.4185.4380.22反应收率/%20.5352.1673.0170.12(2)在镁屑0.3mol(0.76g),硼酸三异丙酯0.10mol(1.88g),反应温度为40℃,反应功率为175w,四氢呋喃用量为20ml,反应时间为100min,滴加时间为30min条件下,2,4-二氟溴苯与硼酸三异丙酯的摩尔比影响如表2所示:表22,4-二氟溴苯:硼酸三异丙酯摩尔比1:11.25:11.5:11.75:1产物质量/g0.871.181.341.06产物纯度/%73.4376.1685.4320.28反应收率/%40.4356.8873.0112.84(3)在镁屑0.3mol(0.76g),2,4-二氟溴苯0.15mol(1.92g),硼酸三异丙酯0.10mol(1.88g),反应功率为175w,四氢呋喃用量为20ml,反应时间为100min,滴加时间为30min条件下,反应温度的影响如表3所示:表3反应温度/℃1020304050产物质量/g0.90.961.171.340.97产物纯度/%68.6377.8666.7385.4372.76反应收率/%39.0947.3156.7473.0144.67(4)在镁屑0.3mol(0.76g),2,4-二氟溴苯0.15mol(1.92g),硼酸三异丙酯0.10mol(1.88g),反应温度为40℃,四氢呋喃用量为20ml,反应时间为100min,滴加时间为30min条件下,超声功率的影响如表4所示:表4反应功率/w125150175200225产物质量/g1.151.041.341.571.32产物纯度/%79.9579.7385.4351.1065.48反应收率/%58.1952.5073.0151.7051.36(5)在镁屑0.3mol(0.76g),2,4-二氟溴苯0.15mol(1.92g),硼酸三异丙酯0.10mol(1.88g),反应功率为175w,反应温度为40℃,反应时间为100min,滴加时间为30min条件下,醚类溶剂的用量和种类的影响如表5所示:表5四氢呋喃用量/ml及种类1020304020(methf)产物质量/g1.221.341.081.091.09产物纯度/%81.8985.4370.0577.0662.80反应收率/%63.2373.0147.8853.1642.32(6)在镁屑0.3mol(0.76g),2,4-二氟溴苯0.15mol(1.92g),硼酸三异丙酯0.10mol(1.88g),反应功率为175w,反应温度为40℃,四氢呋喃用量为20ml,滴加时间为30min条件下,反应时间的影响如表6所示:表6反应时间/min406080100120产物质量/g1.131.051.131.341.17产物纯度/%72.8976.3782.3185.4380.60反应收率/%52.1350.7558.8773.0159.69(7)在镁屑0.3mol(0.76g),2,4-二氟溴苯0.15mol(1.92g),硼酸三异丙酯0.10mol(1.88g),反应功率为175w,反应温度为40℃,四氢呋喃用量为20ml,反应时间为100min条件下,滴加时间的影响如表7所示:表7滴加时间/min010305070产物质量/g1.431.171.341.150.80产物纯度/%44.6973.4885.4364.5280.09反应收率/%40.4554.4173.0146.9643.08实施例4(1)取一个100ml圆底烧瓶,一个60ml恒压滴定漏斗,取0.3mol(0.76g)镁屑、0.10mol(1.88g)硼酸三异丙酯加入圆底烧瓶,取0.15mol(2.88g)2,4-二氟溴苯、20ml四氢呋喃加入恒压滴定漏斗,边滴加超声,超声功率175w,反应温度40℃,30min滴完,滴完后继续反应70min。反应结束后,滤除未反应的镁屑,向滤液中加入5ml0.1mol/l盐酸,溶液将出现白色絮状物,继续添加5ml0.1mol/l的盐酸,絮状物消失,溶液变澄清,用20ml乙酸乙酯萃取分液,有机相用15ml去离子水洗涤一次,水相用乙酸乙酯萃取2×10ml,然后合并有机相,在55℃下旋蒸30min得1.30g粗产物。经液相色谱检测产品纯度为86.36%,产品收率为71.05%。(2)取所制得粗产品1.30g溶于10ml乙酸乙酯,再加入0.1mol/lnaoh溶液6ml,调节ph至10,萃取分液,并用去离子水洗两次油相,合并水相,用0.5mol/l的盐酸调节水相的ph至1,用乙酸乙酯萃取水相,分液,取油相旋蒸,得白色固体1.05g,经液相色谱检测纯度97.89%,两步收率为65.05%。实施例5(1)取一个100ml圆底烧瓶,一个60ml恒压滴定漏斗,取0.3mol(0.76g)镁屑、0.10mol(1.88g)硼酸三异丙酯加入圆底烧瓶,取0.15mol(2.88g)2,4-二氟溴苯、20ml四氢呋喃加入恒压滴定漏斗,边滴加超声,超声功率175w,反应温度40℃,30min滴完,滴完后继续反应70min。反应结束后,滤除未反应的镁屑,向滤液中加入5ml0.1mol/l盐酸,溶液将出现白色絮状物,继续添加5ml0.1mol/l的盐酸,絮状物消失,溶液变澄清,用20ml乙酸乙酯萃取分液,有机相用15ml去离子水洗涤一次,水相用乙酸乙酯萃取2×10ml,然后合并有机相,在55℃下旋蒸30min得1.40g粗产物。经液相色谱检测产品纯度为80.84%,产品收率为71.63%。(2)取制得粗产品1.40g溶于10ml乙酸乙酯,再加入0.1mol/lnaoh溶液6ml,调节ph至13,萃取分液,并用去离子水洗两次油相,合并水相,用0.5mol/l的盐酸调节水相的ph至3,用乙酸乙酯萃取水相,分液,取油相旋蒸,得白色固体1.09g,经液相色谱检测纯度98.99%,两步收率为68.29%。实施例6(1)取一个100ml圆底烧瓶,一个60ml恒压滴定漏斗,取0.3mol(0.76g)镁屑、0.10mol(1.88g)硼酸三异丙酯加入圆底烧瓶,取0.15mol(2.88g)2,4-二氟溴苯、20ml四氢呋喃加入恒压滴定漏斗,边滴加超声,超声功率175w,反应温度40℃,30min滴完,滴完后继续反应70min。反应结束后,滤除未反应的镁屑,向滤液中加入5ml0.1mol/l盐酸,溶液将出现白色絮状物,继续添加5ml0.1mol/l的盐酸,絮状物消失,溶液变澄清,用20ml乙酸乙酯萃取分液,有机相用15ml去离子水洗涤一次,水相用乙酸乙酯萃取2×10ml,然后合并有机相,在55℃下旋蒸30min得1.34g粗产物。经液相色谱检测产品纯度为86%,产品收率为73%。(2)取制得粗产品1.34g溶于10ml乙酸乙酯,再加入0.1mol/lnaoh溶液6ml,调节ph至13,萃取分液,并用去离子水洗两次油相,合并水相,用0.5mol/l的盐酸调节水相的ph至1,用乙酸乙酯萃取水相,分液,取油相旋蒸,得白色固体1.15g,经液相色谱检测纯度96%,两步收率为70%。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1