乳酸左氧氟沙星新晶型及其制备方法和应用与流程
本发明属于医药技术领域,具体涉及乳酸左氧氟沙星的晶型及其制备方法和应用,尤其涉及稳定的乳酸左氧氟沙星一水合物及其制备方法和应用。
背景技术:
喹诺酮类抗菌药是一类以4-喹诺酮为母核的抗菌药,为20世纪90年代人工合成,喹诺酮类抗菌药物通过与dna、dna促旋酶或拓扑异构酶ⅳ发生交叉作用形成三元复合物,诱导dna和拓扑异构酶ⅳ发生构型改变,从而使酶对dna不能产生正常功能,最后导致dna降解及菌体死亡。氟喹诺酮类药物中最畅销的产品是环丙沙星和左氧氟沙星,二者占据了该类药物全球市场的65%。左氧氟沙星为氧氟沙星的左旋异构体,其体外抗菌活性是氧氟沙星的2倍,是右旋体的8~128倍,具有高效、广谱、安全等特点,是目前最具有发展前景的氟喹诺酮类药物之一。
左氧氟沙星,化学名(s)-(-)-9-氟-2,3-二氢-3-甲基-10-(4-甲基-1-哌嗪基)-7-氧-7h吡啶[1,2,3-de]-[1,4]苯并噁嗪-6-羧酸,包括无水左氧氟沙星和左氧氟沙星水合物,无水左氧氟沙星存在多晶型现象(a、b、c、f、g、h、α、β、γ);而左氧氟沙星水合物也存在着多晶现象,包括半水合物和一水合物。
左氧氟沙星在水中的溶解度较小,很难满足临床需求,目前临床上主要应用的是其无机酸盐(盐酸盐)和有机酸盐(乳酸盐)等。cai-yunwang等人在jthermanalcalorim(2013)111:959–963的公开文献中描述了乳酸左氧氟沙星对大肠杆菌的体外抑菌活性优于盐酸左氧氟沙星。
cn102746320a首先公开了盐酸左氧氟沙星的三种晶型,包括一水合物晶型(晶型i)、无水晶型(晶型ii)和半水合物晶型(晶型iii),并在专利中公开了无水晶型(晶型ii)的具体晶胞参数,明确了晶型ii的最小不对称单元中仅包含2个盐酸左氧氟沙星分子。
专利cn106632036a中公开了乳酸环丙沙星的制备方法。m.d.prasanna等人在journalofmolecularstructure(2001)559:255–261的公开文献中描述了乳酸环丙沙星的晶体结构,明确了乳酸环丙沙星中含有1.5个水分子。
但是,迄今为止,尚未有相关专利、文献对乳酸左氧氟沙星晶型进行研究报道。中国药典2004版中未指出乳酸左氧氟沙星存在多晶型现象,仅在结构式中体现出为半水合物,乳酸左氧氟沙星半水合物的理论水含量为1.95%,然而水份测试项中规定为不得超过6%,这与理论值的水含量相差较大,也暗示了乳酸左氧氟沙星半水合物具有较大的吸湿性。
多晶型是不同的固体形式,其享有构成晶体化合物的分子式,但是可能具有不同的物理和化学性质,而这些性质可能会对机械性质、稳定性、吸湿性、溶解性、形态学和生物利用度,以及其他的重要的药物特性产生益处或弊端。
技术实现要素:
本发明所要解决的技术问题在于提供一种乳酸左氧氟沙星的新晶型及其制备方法和应用。本发明的乳酸左氧氟沙星新晶型是含有一个结晶水的乳酸左氧氟沙星,其纯度高,易于溶解、澄清度好、晶习完整、过滤性佳、流动性好,存储过程中吸湿性小,稳定好,可以长期贮存。本发明的制备方法选用溶剂安全价廉易得,反应条件温和,产物收率高于90%,且得到的晶体纯度高(包括晶型纯度和化学纯度),成品的化学纯度可达到99.9%以上,含左氧氟沙星可达到80.0%以上(按无水物计算)。
为解决上述技术问题,本发明提供的技术方案如下。
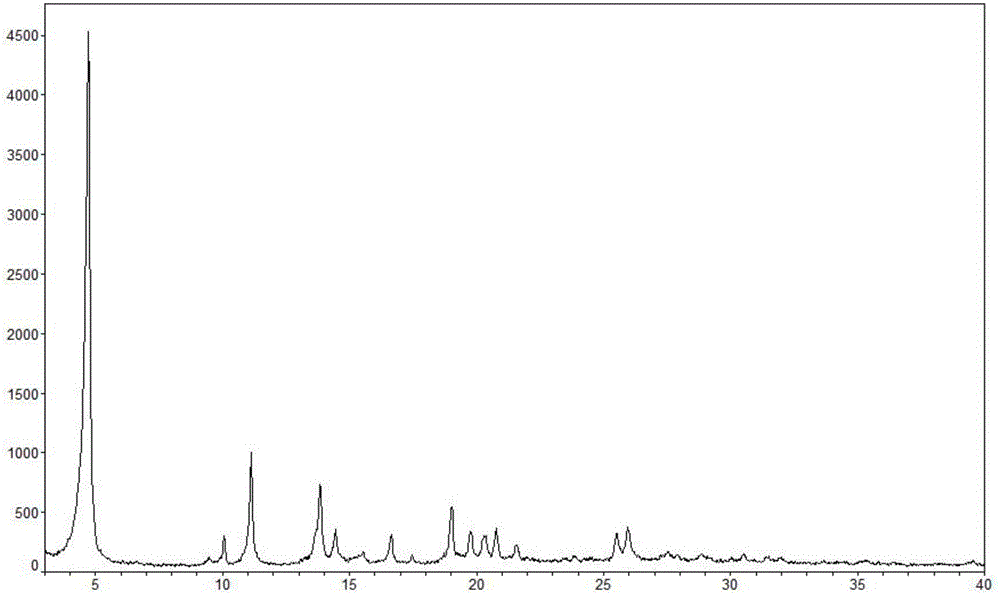
本发明提供了一种乳酸左氧氟沙星一水合物晶型,其在使用cu-kα射线进行测定的x-射线粉末衍射图谱中,至少在下述衍射角2θ为4.72±0.1°、9.44±0.1°、10.06±0.1°、11.12±0.1°、13.82±0.1°、14.44±0.1°、15.52±0.1°、16.62±0.1°、17.46±0.1°、19.00±0.1°、19.74±0.1°、20.34±0.1°、20.74±0.1°、21.52±0.1°、25.48±0.1°和25.92±0.1°处有特征峰。
较佳地,所述乳酸左氧氟沙星一水合物晶型的的x-射线粉末衍射图的特征衍射谱线如后文表2所示。
更佳地,所述乳酸左氧氟沙星一水合物晶型的x-射线粉末衍射图谱基本上如图1所示。
进一步地,乳酸左氧氟沙星一水合物晶型可以通过x-射线单晶衍射分析来表征。其最小不对称单元由1个质子化左氧氟沙星分子、1个乳酸分子和1个水分子构成,如图2所示。所述乳酸左氧氟沙星一水合物的晶体属于单斜晶系(monoclinic),c2空间群,晶胞参数为
进一步地,乳酸左氧氟沙星一水合物晶型可以通过热分析来表征。其典型的dsc图在图4中显示。dsc图中,包含在30℃~120℃有三个吸热峰(峰顶值分别约52℃、63℃、92℃),为脱水吸热峰。本发明提供的乳酸左氧氟沙星一水合物晶型,理论含水量为3.83%,tga显示从室温至120℃范围内,其失重率约为3.9-4.3%,证明其结构中含有一个水分子。其典型的tga图在图3中显示。
本发明还提供了所述乳酸左氧氟沙星一水合物晶型的制备方法,其包括下述步骤:
1)将左氧氟沙星或乳酸左氧氟沙星,和溶剂混合,搅拌并升温至回流;
2)步骤1)原料为左氧氟沙星,向步骤1)所得的混合液中滴加乳酸成盐剂,保温回流搅拌,然后降温析晶;
或者,步骤1)原料为乳酸左氧氟沙星,降温析晶;
3)过滤,洗涤,干燥后,即得;
步骤1)中,所述的溶剂为有机溶剂和水的混合溶剂,所述有机溶剂为醇类溶剂、酮类溶剂、醚类溶剂和酯类溶剂中的一种或多种。
步骤1)中,所述醇类溶剂较佳地为甲醇、乙醇和异丙醇中的一种或多种;所述酮类溶剂较佳地为丙酮和/或甲基乙基酮;所述醚类溶剂较佳地为四氢呋喃;所述酯类溶剂较佳地为乙酸乙酯和/或乙酸异丙酯。在本发明的一优选实施方式中,所述溶剂为质量浓度85%-95%的乙醇溶液。在选用质量浓度85%-95%的乙醇溶液作为溶剂的时候,所述回流温度一般在80-90℃,优选回流温度为85-90℃。
步骤1)中,所述左氧氟沙星和溶剂的用量可按本领域常规方法进行选择,所述左氧氟沙星和溶剂的重量比优选1:(3-10),更优选1:(3-8)。
步骤2)中,所述的乳酸成盐剂可为常规市售的dl-乳酸。所述乳酸成盐剂可以是纯乳酸,或是乳酸溶液,例如质量浓度33%-85%的乳酸溶液,优选质量浓度33%的乳酸溶液。所述乳酸成盐剂的用量可按本领域常规方式进行确定,一般为乳酸的摩尔量是左氧氟沙星摩尔量的0.98-1.02倍。
步骤2)中,所述保温回流搅拌的方法和条件可为本领域常规的方法和条件。所述保温回流的时间较佳地为1-2h。
步骤2)中,所述降温析晶的方法和条件可为本领域常规的方法和条件。
本发明的一优选实施方式中,所述降温析晶的方法为:①先降温进行静置养晶;②静置养晶结束后,再搅拌降温至10℃以下。步骤①中,静置养晶的温度可按本领域常规方法进行选择。在选用质量浓度85%-95%的乙醇溶液作为溶剂的时候,所述养晶的温度较佳地为40-50℃,更佳地为45-50℃。所述养晶的时间较佳地为1-3h。步骤①中,所述降温的方法和条件可为本领域常规的方法和条件,所述降温的速率较佳地为0.5-3℃/min,更佳地为1-2℃/min。步骤②中,所述搅拌的方法和条件可为本领域常规的方法和条件。所述搅拌的速率较佳地为100-150r/min。若搅拌速度过快,晶体的结晶性会有所降低,其过滤性和热稳定性也会下降。步骤②中,所述降温的方法和条件可为本领域常规的方法和条件,所述降温的速率较佳地为0.3-1℃/min,更佳地为0.5-0.8℃/min。
本发明的另一优选实施方式中,所述降温析晶的方法为:搅拌降温至10℃以下。所述降温的方法和条件可为本领域常规的方法和条件。所述降温的速率较佳地为0.3-1℃/min,更佳地为0.5-0.8℃/min
步骤3)中,所述过滤、洗涤和干燥的方法和条件均为本领域常规的方法和条件。所述过滤即是将步骤3)所得混合液中的晶体与液体分离的步骤。按本领域常识,所述酸左氧氟沙星一水合物晶型中含有结晶水,所述干燥的温度不宜高于50℃,一般可在40-50℃环境下进行真空干燥,或进行冷冻干燥。
本发明还提供了所述乳酸左氧氟沙星一水合物晶型在制备抗菌药物中的应用。
本发明中,“晶型”一词不仅理解为“晶体类型”或“晶体结构”;在技术方案中,“晶型”更理解为“具有特定晶体结构的物质”或“特定晶体类型的晶体”。例如,在技术方案中,“乳酸左氧氟沙星一水合物晶型”可以理解为“具有特定晶体结构的乳酸左氧氟沙星一水合物”或“特定晶体类型的乳酸左氧氟沙星一水合物的晶体”。
本发明中,所述晶型均被所示的x射线衍射图表征所证实。本领域技术人员能够理解,其中的实验误差取决于仪器的条件、样品的准备和样品的纯度。特别是,本领域技术人员公知,x射线衍射图通常会随着仪器的条件而有所改变。另外,峰角度的实验误差通常在5%或更少,这些角度的误差也应该被考虑进去,通常允许有±0.1°的误差。另外,由于样品高度等实验因素的影响,会造成峰角度的整体偏移,通常允许一定的偏移。因而,本领域技术人员可以理解的是,任何具有和本发明图谱中的特征峰相同或相似的图的晶型均属于本发明的范畴之内。
本发明中,按本领域常识,dsc测试的误差也被考虑进去,通常峰值的测量允许有±1℃的误差。
本发明中,所述“室温”为本领域常规意义上的室温温度,一般为10~30℃。
在符合本领域常识的基础上,上述各优选条件,可任意组合,即得本发明各较佳实例。
本发明所用试剂和原料均市售可得。
本发明的积极进步效果在于:
(1)本发明的乳酸左氧氟沙星一水合物晶型,其色泽好、易于溶解、澄清度好、晶习完整、过滤性佳、流动性好,存储过程中吸湿性小,稳定好,可以长期贮存。
(2)本发明制备的乳酸左氧氟沙星一水合物晶型的方法,产物收率高于90%;成品的化学纯度可达到99.9%以上,含左氧氟沙星可达到80.0%以上(按无水物计算)。
(3)本发明的制备方法简单,选用溶剂安全价廉易得,反应条件温和,且得到的晶体纯度高(包括晶型纯度和化学纯度)。
附图说明
图1是乳酸左氧氟沙星一水合物晶型的x-射线粉末衍射(pxrd)图(横坐标:衍射角2θ(°);纵坐标:相对强度(count))。
图2是乳酸左氧氟沙星一水合物晶型的ortep(单晶结构分析)图。
图3是乳酸左氧氟沙星一水合物晶型的热重分析(tg)图(横坐标:温度(℃);纵坐标:重量(%))。
图4是乳酸左氧氟沙星一水合物晶型的差示扫描分析(dsc)图(横坐标:温度(℃);纵坐标:热流(w/g))。
图5是乳酸左氧氟沙星半水合物晶型的x-射线粉末衍射(pxrd)图(横坐标:衍射角2θ(°);纵坐标:相对强度(count))。
图6是乳酸左氧氟沙星半水合物晶型的热重分析(tg)图(横坐标:温度(℃);纵坐标:重量(%))。
图7是乳酸左氧氟沙星半水合物晶型的差示扫描分析(dsc)图(横坐标:温度(℃);纵坐标:热流(w/g))。
具体实施方式
下面通过实施例的方式进一步说明本发明,但并不因此将本发明限制在所述的实施例范围之中。下列实施例中未注明具体条件的实验方法,按照常规方法和条件,或按照商品说明书选择。
本发明中检测药物多晶型结构及性能的仪器如下:
单晶衍射:rigakur-axis-rapid单晶衍射仪,采用mokα
粉末x射线衍射(xrd)表征:仪器:rigakud/max-2550pc,cukα辐射,功率40kv×250ma,扫描范围2θ3~40°,步宽(stepwidth)0.02°,扫描速度5°/min。
热重分析(tg)表征:仪器:ta公司sdtq600,吹扫气:氮气120ml/min,升温速度:10℃/min,温度范围:室温~400℃。
差示扫描量热分析(dsc)表征:仪器:ta公司dscq100,吹扫气:氮气50ml/min,升温速度:10℃/min,温度范围:室温~200℃。
下述实施例中,所用的百分比若无特别指出,均为质量百分比。
实施例1
乳酸左氧氟沙星一水合物晶型制备方法如下:
1)将左氧氟沙星10g和质量浓度为95%乙醇32.5g投入三口瓶中,搅拌,升温至80℃;
2)向步骤1)所得的混合液中滴加质量浓度33%的乳酸溶液7.4g,滴加时间为1min,保温搅拌1h,以1℃/min的速率降温至50℃,进行静置养晶2h;静置养晶结束,以100-150r/min速度搅拌,以0.5℃/min的速率降温至8℃;
3)滤取步骤2)所得混合液中的晶体后,用无水乙醇洗涤,然后将晶体在45℃下真空干燥,即得。
该制备方法的收率为94.34%;经pxrd检测,为乳酸左氧氟沙星一水合物晶型;经hplc检测,其质量纯度为99.96%;左氧氟沙星的含量为80.3%;经tga检测,水分3.9%。
对按以上制备方法得到的乳酸左氧氟沙星一水合物晶型,通过单晶培养得到适用于x-射线单晶衍射的完美晶体。单晶培养的具体方法如下:
称取前述制备得到的乳酸左氧氟沙星一水合物晶型0.1g,加入19ml四氢呋喃和1ml水,加热溶解,静置1~3天后得到适用于x-射线单晶衍射的完美晶体。
乳酸左氧氟沙星一水合物晶型的单晶结构分析表明,乳酸左氧氟沙星一水合物晶型属于属单斜晶系(monoclinic),c2空间群,最小不对称单元中含有一个乳酸左氧氟沙星分子和一个水分子,结晶学参数如下:
该乳酸左氧氟沙星一水合物晶型的特征x射线粉末衍射图为图1所示,特征谱线见表2。获得的乳酸左氧氟沙星一水合物晶型的热重分析(见图3)表明其含有一个结晶水。其dsc图在图3中显示,可以看出,包含在30℃~120℃有三个吸热峰(峰顶值分别约52℃、63℃、92℃),为脱水吸热峰。
表1乳酸左氧氟沙星一水合物晶型的原子坐标
表2乳酸左氧氟沙星一水合物晶型的特征衍射谱线
效果实施例1稳定性实验
对本发明的乳酸左氧氟沙星一水合物晶型的稳定性进行了研究,结果见下表3和表4。
表3加速实验(考察条件40±2℃、rh75±5%)
表4长期试验(考察条件25±2℃、rh60±5%)
结果显示,在加速试验、长期试验条件下从0-6个月,外观、澄清度、杂质、水分、含量没有变化,说明该乳酸左氧氟沙星一水合物晶型化学稳定性好,适合制备制剂,可以长期贮存,在医药上有很高的应用价值。
效果实施例2热稳定性实验
众所周知,药物的水合物晶型热稳定性均较差,在干燥过程中往往会发生部分脱水引起转晶。因此,在干燥过程中,干燥的温度和时间对产品的固体形态有着较大的影响。发明人对本发明所述的乳酸左氧氟沙星一水合物晶型进行了热稳定实验。分别于50℃热处理30min、50℃热处理150min、55℃热处理60min、65℃热处理60min、70℃热处理90min、80℃热处理90min、90℃热处理90min后,用pxrd进行检测分析。
结果显示,在50℃及以下进行热处理时,本发明所述的乳酸左氧氟沙星一水合物晶型能稳定存在。在大于50℃且小于70℃的温度下进行热处理时,开始出现微小的相变;在70℃以上进行热处理时,开始出现明显的相变,说明本发明所述的乳酸左氧氟沙星一水合物晶型热稳定性较好。
实施例2
乳酸左氧氟沙星一水合物晶型制备方法如下:
1)将左氧氟沙星10g和质量浓度为95%乙醇50g投入三口瓶中,搅拌,升温至80℃;
2)向步骤1)所得的混合液中滴加33%乳酸溶液7.4g,滴加时间为2min,保温搅拌1.5h,以1℃/min的速率降温至50℃,进行静置养晶2h;静置养晶结束,以100-150r/min速度搅拌,以0.5℃/min的速率降温至6℃,
3)滤取步骤2)所得混合液中的晶体后,用无水乙醇洗涤,然后将晶体在45℃下真空干燥,即得。
该制备方法的收率为91.69%;经pxrd检测,为本发明的乳酸左氧氟沙星一水合物晶型;经hplc检测,其质量纯度为99.97%;左氧氟沙星的含量为80.4%;经tga检测,水分4.0%。
实施例3
乳酸左氧氟沙星一水合物晶型制备方法如下:
1)将左氧氟沙星10g和质量浓度为90%乙醇50g投入三口瓶中,搅拌,升温至80℃;
2)向步骤1)所得的混合液中滴加33%乳酸溶液7.4g,滴加时间为2min,保温搅拌1.5h,以1℃/min的速率降温至50℃,进行静置养晶2h;静置养晶结束,以100-150r/min速度搅拌,以0.5℃/min的速率降温至6℃,
3)滤取步骤2)所得混合液中的晶体后,用无水乙醇洗涤,然后将晶体在50℃下真空干燥,即得。
该制备方法的收率为90.82%;经pxrd检测,为本发明的乳酸左氧氟沙星一水合物晶型;经hplc检测,其质量纯度为99.96%;左氧氟沙星的含量为80.3%;经tga检测,水分4.3%。
实施例4
乳酸左氧氟沙星一水合物晶型制备方法如下:
称取左氧氟沙星10g,投入三口瓶中,加入80g水饱和乙酸乙酯,,搅拌,升温至80℃;滴加质量浓度33%的乳酸溶液7.4g,滴加时间为1min,保温搅拌1h,以0.5℃/min的速率降温至8℃,滤取混合液中的晶体后,用乙酸乙酯洗涤,然后将晶体在45℃下真空干燥,即得。经pxrd检测,为本发明的乳酸左氧氟沙星一水合物晶型。
实施例5
乳酸左氧氟沙星一水合物晶型制备方法如下:
称取乳酸左氧氟沙星10g,投入三口瓶中,加入50g的95%丙酮-水溶液,搅拌,升温至回流,直至完全溶解,以0.5℃/min的速率降温至5℃,滤取混合液中的晶体后,用丙酮洗涤,然后将晶体在45℃下真空干燥,即得。经pxrd检测,为本发明的乳酸左氧氟沙星一水合物晶型。
对比实施例
乳酸左氧氟沙星半水合物制备方法如下:
称取乳酸左氧氟沙星一水合物1g,投入三口瓶中,加入20ml二甲基亚砜,升温至50℃,搅拌,直至完全溶解。快速冷却至10℃析晶。过滤,得到固体粉末。pxrd检测谱图,如图5所示,特征衍射谱线见表5。在室温~110℃范围内的失重率为2.6%(理论失重2.0%),如图6所示;dsc图显示其在92.6℃(峰顶值)处有一吸热峰,为脱去半个水所引起的相变吸热,熔融分解温度为210.8℃,如图7所示。
表5乳酸左氧氟沙星半水合物晶型的特征衍射谱线
- 还没有人留言评论。精彩留言会获得点赞!